Неорганика. Закон сталості cкладу
 Скачать 0.55 Mb. Скачать 0.55 Mb.
|

Тиск пари розчинівРозчинена речовина ускладнює випаровування розчинника внаслідок взаємодії молекул речовини і розчинника. Тому при постійній температурі тиск пари над розчином нелеткої речовини менше, ніж над розчинником, за винятком розчинів, в яких речовина є леткою (NH3, HCl). Зниження тиску пари над розчином тим більше, чим вища концентрація розчину. Залежність зниження тиску пари розчинів від їх концентрації виражається першим законом Рауля: відносне зниження тиску насиченої пари над розчином прямо пропорційне мольній частці розчиненої речовини. Математично цей закон Рауля можна записати так: де р0 – тиск пари чистого розчинника; р – тиск пари розчину; N – число молів розчинника; n – число молів розчиненої речовини; p = p0 p/p– p – зниження тиску пари; 0 – відносне зниження тиску пари над розчином; n/(N+n) – мольна частка розчиненої речовини. Враховуючи ці позначення, закон Рауля можна записати у такому вигляді: p = p0n/(N+n). Для розбавлених розчинів, коли n«N, закон можна спростити: p = р0n/N. З рівняння витікає, що як і у разі осмотичного тиску, зниження тиску пари не залежить від природи розчиненої речовини, а обумовлено тільки числом його молів в певній кількості розчинника, тобто його концентрацією. ^ Температура кипіння і температура замерзання розчинів Температура кипіння і температура замерзання розчинів залежать від тиску пари розчинів. Зниження пари розчину викликає підвищення температури кипіння або зниження температури замерзання розчину порівняно з відповідними температурами для чистого розчинника. Рауль показав (другий закон), що tпідвищення температури кипіння (кипt) або зниження температури замерзання (зам) розчину прямо пропорційне моляльній концентрації розчиненої речовини, або tкип = kебСm; tзам = kкрСm, де Сm – моляльна концентрація розчину; kеб і kкр – коефіцієнти пропорційності, ебуліоскопічна і кріоскопічна сталі. Зако́н Рау́ля (рос. закон Рауля; англ. Raoult’s law; нім. Raoultsches Gesetz n) — парціальні тиски рі пари кожного з компонентів ідеального розчину при постійній температурі є пропорційними до молярних часток цих компонентів в рідкій фазі де Закон установив Франсуа Марі Рауль. Залежність зниження тиску пари розчинів від їхньої концентрації виражається першим законом Рауля (1887): відносне зниження тиску насиченої пари над розчином прямопропорційне мольній частці розчиненої речовини. Другий закон Рауля: підвищення температури кипіння, або зниження температури замерзання розчину прямопропорційне моляльнійконцентрації розчиненої речовини. Коефіцієнти пропорційності називають відповідно ебуліоскопічна і кріоскопічна сталі. Значення коефіцієнтів залежать лише від природи розчинника. Застосування На основі закону Рауля можна визначати молекулярну масу речовин (неелектролітів). Розчини електролітів не підлягають закону Рауля внаслідок електролітичної дисоціації (через збільшення кількості часточок у розчині). ТЕМПЕРАТУРИ ЗАМЕРЗАННЯ ТА КИПІННЯ РОЗЧИНІВ Зниження тиску пари над розчином впливає на температуру замерзання (кристалізації) та кипіння розчинів. Зменшення тиску пари розчину спричинює підвищення температури кипінняабо зниження температури замерзаннярозчину порівняно з відповідними температурами для чистого розчинника (див. рис. З.6.). Згідно з другим законом Рауля(висновок з першого закону): підвищення температури кипіння та зниження температури замерзання розчину відносно чистого розчинника пропорційні молярній концентрації розчиненої речовини: де Сm — моляльність розчину; Е і К— ебуліоскопічната кріоскопічна сталі,які залежать лише від властивостей розчинника — його температури кипіння (замерзання) Тк(3), молярної маси М і теплоти випаровування (плавлення) ΔНвип(пл), але не залежать від властивостей розчиненої речовини. Наприклад, для води ебуліоскопічна стала Аналогічно визначають і кріоскопічну сталу. Ебуліоскопічна та кріоскопічна сталі вказують на підвищення температури кипіння або зниження температури замерзання розчину з концентрацією Ст= 1 моль/кг. Оскільки для таких концентрованих розчинів рівняння не придатні, то сталі Е і К є екстраполяційними, тобто їх визначають експериментально для розведених розчинів із перерахунком на один моль розчиненої речовини. На зміні температур кипіння та замерзання розчинів базуються ебуліоскопічнийі кріоскопічний методи визначення молекулярних масречовин де т1і т2 — маси розчиненої речовини та розчинника; М— молярна маса розчиненої речовини. Пунктиром показані залежності тисків компонентів від складу для ідеальних розчинів. КОНЦЕНТРАЦІЯ РОЗЧИНІВ—це величина, яку вимірюють масою або об’ємом розчиненої речовини, що міститься у певній масі або об’ємі розчину або розчинника. Найчастіше використовують такі способи вираження концентрації: масова частка, молярна, моляльна концентрація, молярна концентрація еквівалента, молярна частка, об’ємна частка та титр. Масову частку речовини Х w(Х) розраховують за формулою: де m(X) — маса розчиненої речовини Х (в кг, г); m – маса розчину (в кг, г). Її виражають у частках одиниці або в %. В останньому випадку розрахунок здійснюють за формулою: 13. Теорія електролітичної дисоціації, основні положення. ТЕОРІЯ ЕЛЕКТРОЛІТИЧНОЇ ДИСОЦІАЦІЇ Властивості електролітів узагальнені основоположником теорії електролітичної дисоціації Арреніусом (1887) і розвинуті у працях Кістяківського та Каблукова на основі хімічної теорії розчинів Менделєєва. В основу теорії електролітичної дисоціаціїпокладено такі положення: розчинення електроліту супроводжується розпадом його молекул на іони; електролітична дисоціація є оборотним процесом; електропровідність розчинів електролітів пропорційна загальній концентрації іонів у розчині. Арреніус вважав, що молекули електролітів розпадаються на іони не під дією електричного струму, а в процесі розчинення, хоч його теорія не враховувала всієї складності явищ у розчинах. Зокрема, іони вважалися вільними частинками, що не залежали від молекул розчинника. Згідно зі сучасним поглядом, розпад полярних молекул (іонізація)або іонних кристалів(дисоціація)розчиненої речовини на іони відбувається під впливом полярних молекул розчинника, внаслідок чого утворюються хімічні сполуки — сольвати, а коли розчинником є вода — гідрати. Процес гідратації (сольватації) екзотермічний і відбувається спонтанно зі зменшенням ентальпії. Одночасно з дисоціацією відбувається й зворотний процес — асоціація. Великі за розміром гідратні (сольватні) оболонки іонів заважають останнім при їх зіткненні у розведених розчинах знову утворювати молекули (кристали) вихідних речовин. Взаємодія між протилежно зарядженими іонами у розчині послаблюється також під впливом розчинників. Мірою такого впливу єдіелектрична проникністьрозчинника. Чимбільше значення діелектричної проникності середовища, тим краще дисоціює електроліт (правило Нернста, 1894): де F—сила взаємодії між іонами; zК і zа — заряди катіона та аніона; e — заряд електрона; NA — число Авогадро; έ — діелектрична проникність середовища; r- віддаль між центрами іонів. Енергія електростатичної взаємодії у воді у 81 раз менша, ніж у вакуумі, тому у воді електроліти дисоціюють значно ліпше, ніж у розчинниках з малими значеннями є. У бензолі та інших подібних розчинниках електроліти майже не дисоціюють. Проте правило Нернста має винятки. Кількісною характеристикою повноти перебігу електролітичної дисоціації є ступінь дисоціації α— відношення числа частинок електроліту, що розпалися на іони, до загального числа його частинок у розчині: Ступінь дисоціації залежить від природи електроліту та розчинника, від концентрації електроліту й температури розчину. Залежно від значення α електроліти умовно поділяють на сильні, слабкіта середні.До сильних електролітівналежать майже всі солі, гідроксиди лужних і лужноземельних металів, багато кислот (НС1, НNОз, НСlO4, Н2SО4 тощо), до слабких— більшість основ, амфотерні гідроксиди, деякі кислоти (Н2S, Н3ВО3, Н2СО3, НСN, Н2SiO3, НNO2). Поділ електролітів на сильні та слабкі є умовним, оскільки сила електроліту значно залежить від природи розчинника, його діелектричної проникності. Насправді електролітом є не сама розчинена речовина, а речовина разом з розчинником (електролітичний розчин).Наприклад, водний розчин хлороводню є сильним електролітом, але бензольний розчин хлороводню практично не проводить електричний струм. Ступінь дисоціації визначають експериментально за значенням ізотонічного коефіцієнта або вимірюванням електропровідності розчинів різних концентрацій. 14. Ступінь дисоціації , сильні та слабкі електроліти. Сильні та слабкі електроліти Усі електроліти поділяють на сильні та слабкі. Сильні електроліти в розчинах дисоціюють повністю, а слабкі — частково, тобто частина молекул залишається в недисоційованому стані. До сильних електролітів відносять усі солі (за незначним винятком), луги (гідроксиди лужних металів, а також Барію, Стронцію й Кальцію) та деякі кислоти (НСl, НВr, НІ, HNO3, H2SO4(розб)). Інші електроліти відносять до слабких. Силу електролітів можна пояснити ступенем іонності зв’язку, що підлягає розриву при дисоціації. Оскільки в солях між іонами металів та кислотних залишків зв’язок іонний, то майже всі солі належать до сильних електролітів. Аналогічний підхід справедливий також і для кислот та основ. Але якщо в основ полярність зв’язку між атомом металу й гідроксильною групою визначається тільки електронегативністю атома металу, то для кислот полярність зв’язку між атомами Оксигену та Гідрогену залежить від якісного й кількісного складу кислотного залишку. У кислот, до складу яких атоми Оксигену не входять НЕ, сила кислот залежить від розміру атома Е. Чим більший радіус атома, тим більша довжина зв’язку Н-Е, а отже, тим простіше його розірвати й тим більшою є сила кислоти. Таким чином, у ряді галогеноводневих кислот зі збільшенням порядкового номера галогену сила кислоти збільшується: плавикова кислота є слабкою, а йодоводнева кислота — сильною, тобто сила кислот змінюється в ряді: HF < НСl < НВr < НІ. Силу оксигеновмісних кислот можна визначити за формулою Е(ОН)mОn. Якщо n < 2 — кислота слабка, якщо n більше або дорівнює 2 — сильна. Взаємозв’язок сили кислоти із числом атомів Оксигену, які не входять до складу гідроксильних груп, можна пояснити в такий, спосіб. Атом Оксигену, як найбільш електронегативний, притягує до себе спільні електронні пари. У результаті електронна густина від атома Оксигену в групі ОН зміщується в бік кислото-твірного елемента й зв’язок між атомами Оксигену та Гідрогену в гідроксильній групі стає більш полярним. Чим більше число атомів Оксигену, які не входять до групи ОН, тим полярніший зв’язок і сильніша кислота. Ступінь дисоціації Для кількісного опису сили електролітів використовують поняття «ступінь дисоціації». Ступенем дисоціації а називають відношення числа молекул, що розпалися на іони (продисоційованих), до загального числа розчинених молекул. 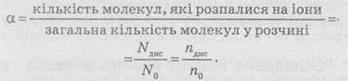 Різні електроліти дисоціюють на іони різною мірою. Як випливає з наведеного рівняння, ступінь дисоціації може змінюватися від 0 до 1. Ступінь дисоціації часто виражають у відсотках. Слід зазначити, що при дисоціації відбувається збільшення числа частинок у розчині. Наприклад, якщо в розчині перебуває електроліт типу Kat+An”, який повністю розпадається на іони, то число частинок збільшується у два рази. Ступінь дисоціації також є характеристикою, за якою можна визначити, є електроліт сильним чи слабким. Так, якщо ступінь дисоціації електроліту в 0,1 М розчині більший 30 %, то його відносять до сильних електролітів, а якщо менший 3% — до слабких. Електроліти, в яких ступінь дисоціації має проміжні значення, відносять до електролітів середньої сили. Ступені дисоціації слабкого електроліту, визначені різними способами (наприклад, за виміром електропровідності або температурою кипіння розчину), найчастіше збігаються. Однак для сильних електролітів визначення ступеня дисоціації різними способами дає різні результати. Отже, величина ступеня дисоціації не характеризує реальну (справжню) ступінь дисоціації, а представляє уявну величину. Цей, а також низка інших фактів призвели до перегляду теорії Арреніуса у 20-х роках минулого сторіччя й до уточнення, внаслідок чого з’явилася нова теорія електролітів, яка враховує електростатичну взаємодію між іонами. Найбільший внесок у розвиток нової теорії зробив голландський учений П. Дебай. Згідно із цією теорією, передбачається, що сильні електроліти в розведених розчинах дисоціюються повністю (α = 1). Відмінність виміряного ступеня дисоціації сильних електролітів від одиниці пояснюється електростатичними взаємодіями між різнойменно зарядженими іонами, тобто, якщо для якого-небудь сильного електроліту виміряний ступінь дисоціації α = 70%, то, згідно із новою теорією електролітів, це означає, що всі молекули дисоційовані на іони, але іони вільні лише на 70%, решта 30% іонів «зв’язані» електростатичними взаємодіями. 15. Реакції іонного обміну, умови реакції. Іонні рівняння. Іонні рівняння реакцій Оскільки електроліти в розчинах утворюють іони, то для відображення змісту реакцій часто використовують так звані іонні рівняння, тому що в розчинах відбуваються реакції не між молекулами, а між іонами. Реакції, чиїм змістом є обмін іонами між реагентами, називають реакціями іонного обміну. У цьому розділі будуть розглядатися тільки реакції обміну в розчинах, що відбуваються без зміни ступеня окиснення елементів. Складання іонних рівнянь реакцій При складанні іонних рівнянь реакцій слід дотримуватися такого алгоритму: 1. Скласти молекулярне рівняння реакції (усі речовини — реагенти й продукти — записують у вигляді молекул) і розставити в ньому коефіцієнти: 2. Скласти повне іонне рівняння реакції. Для цього замість запису молекул усіх речовин, які є сильними електролітами, слід записати іони, у вигляді яких вони існують у розчинах, з урахуванням розставлених коефіцієнтів. Так, у даному разі сильними електролітами є всі речовини, окрім води. Тому замість молекули сульфатної кислоти записуємо іони, на які вона дисоціює в розчині, тобто Н+ і SO42-, замість калій гідроксиду — К+ та ОН-, замість калій сульфату — К+ і SO42-. Вода є дуже слабким електролітом, тому її записують у молекулярній формі. Одержуємо: 3. Скласти скорочене іонне рівняння реакції (іноді його називають іонно-молекулярним рівнянням реакції). Для цього в повному іонному рівнянні необхідно скоротити в лівій і правій частинах рівняння однакові іони. У нашому випадку однаковими є іони SO42- та К+. Одержуємо: У разі потреби можна ще скоротити кратні коефіцієнти в рівнянні: Умови протікання реакцій обміну в розчинах Протилежно заряджені іони в розчинах притягуються й можуть утворювати продукти реакції. Згідно із теорією дисоціації, можливі два варіанти протікання реакцій обміну в розчинах: а) утворювані речовини — сильні електроліти, добре розчинні у воді й повністю дисоціюються на іони; б) одна (або кілька) з утворюваних речовин — газ, осад (речовина, яка погано розчиняється у воді) або слабкий електроліт (добре розчинний у воді). Розгляньмо перший випадок — взаємодію натрій хлориду та калій сульфату — і запишімо молекулярне та іонне рівняння із цими речовинами: І натрій сульфат, і калій хлорид є добре розчинними сполуками й сильними електролітами, тому в цьому разі при складанні скороченого іонного рівняння всі іони скорочуються. Це свідчить про те, що при змішуванні розчинів натрій хлориду та калій сульфату реакція не відбувається, а утворюється суміш іонів Na+, К+, Сl- і SO42-. Другий випадок розгляньмо на прикладі двох реакцій: У іонній формі рівняння (1) та (2) матимуть такий вигляд: 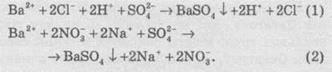 Барій сульфат є практично нерозчинним і вилучається зі сфери реакції, тому його записують у молекулярній формі. Однакові іони в обох частинах рівнянь скорочуються, й обидва рівняння перетворюються на однакові скорочені іонні рівняння: Очевидно, що при взаємодії інших сульфатів з будь-якою розчинною сіллю Барію реакція буде описуватися таким самим скороченим іонним рівнянням. Отже, іонне рівняння, на відміну від молекулярного, належить не до однієї якої-небудь реакції між конкретними речовинами, а до цілої групи аналогічних реакцій. Таким чином, якщо в результаті реакції одна з речовин вилучається зі сфери реакції, то реакція обміну відбувається повністю. Це можливо, якщо одним із продуктів реакції є: 1) слабко-розчинний газ, який виділяється з розчину; 2) слабко-розчинна речовина, що випадає в осад; 3) слабко-дисоційована речовина (слабкий електроліт) — слабкі кислоти, вода. У іншому разі реакція не відбувається, а утворюється суміш іонів. 16. Дисоціація води. Водневий показник (рН середовища) Дисоціація води.pH Водa і як слабкий електроліт незначною мірою дисоціює на іони Н+ і ОН-, що перебувають у рівновазі з недисоційованими молекулами: Н2O ⇆ Н+ + ОН-. Концентрацію іонів звичайно виражають у молях іонів в 1 л. Як видно з рівняння дисоціації води, в ній величини [Н+] і [ОН-] однакові. Експериментально встановлено, що в одному літрі води за кімнатної температури (22 °С) дисоціації піддається лише 10-7 моль води і при цьому утворюється 10 моль/л іонів Н+ і 10-7 моль/л іонів ОН- . Добуток концентрації іонів гідрогену і гідроксид-іонів у воді називається іонним добутком води (позначається Kв). За певної температури Кв — величина стала, чисельно дорівнює при 22 °С 10-14 : Kв = [Н+] [ОН-] = 10-7 • 10-7 = 10-14. (5.4) Сталість добутку [Н+] [ОН-] означає, що в будь-якому водному розчині ні концентрація іонів гідрогену, ні концентрація гідроксид-іонів не може дорівнювати нулю. Іншими словами, бyдь-який водний розчин кислоти, основи або солі містить як Н+, так і ОН- -іони. Дійсно, для чистої води [Н+] = [OН-] = 10-7 моль/л. Якщо до води додати кислоту, то [Н+] стане більшою ніж 10-7, а [ОН- ] — меншою ніж 10-7 моль/л. Навпаки, якщо до води додати луги, то [Н+] стає меншою ніж 10-7 , а [ОН-] — більшою ніж 10-7 моль/л. Зі сталості добутку [Н+] [ОН-] випливає, що при збільшенні концентрації одного з іонів води відповідно зменшується концентрація іншого іона. Це дає змогу обчислити концентрацію Н+ -іонів, якщо відома концентрація ОН- -іонів, і навпаки. Наприклад, якщо у водному розчині [Н+] = 10-3 моль/л, то [ОН-] визначатиметься так: [ОН-] = Kв/[Н+] = 10-14/10-3 = 10-11 моль/л. Отже, кислотність і основність розчину можна виражати через концентрацію або іонів Н+, або іонів ОН-. На практиці користуються першим способом. Тоді для нейтрального розчину [Н+] = 10—7 моль/л, для кислого [Н+] > 10 -7, для лужного [Н+] < 10-7 моль/л. Щоб уникнути незручностей, пов’язаних із застосуванням чисел з від’ємними показниками ступеня, концентрацію іонів гідрогену прийнято виражати через водневий показник, що позначається символомpH (читається “пе-аш”). Водневим показником pH називається десятковий логарифм концентрації водневих іонів, взятий з протилежним знаком: pH = -lg[Н+] (5.5) або [Н+] = 10-рН, (5.6) де [Н+] — концентрація іонів гідрогену, моль/л. Поняття “водневий показник” введене датським хіміком С. Серенсеном у 1909 p.: літера “р” — початкова літера датського слова potenz — математичний ступінь, буква “Н” — символ гідрогену. За допомогою pH реакція розчинів характеризується так: нейтральна — pH =7, кисла — pH < 7, лужна — pH > 7. Наведемо значення pH деяких найвідоміших розчинів і зазначимо відповідну їм реакцію середовища: шлунковий сік — pH = 1,7 (сильнокисла реакція), торф’яна вода — pH = 4 (слабкокисла), дощова вода — pH = 6 (слабкокисла), вода з водопроводу — pH = 7,5 (слабколужна), кров — pH = 7,4 (слабколужна), слина — pH = 6,9 (слабкокисла), сльози — pH = 7 (нейтральна). Винятково значна роль pH в найрізноманітніших явищах і процесах — і в природі, і в техніці. Багато виробничих процесів у хімічній, харчовій, текстильній та інших галузях промисловості відбуваються лише за певної реакції середовища. Так само необхідна для нормального розвитку сільськогосподарських культур і одержання високих урожаїв і певна реакція ґрунтового розчину. Залежно від значення pH ґрунтового розчину ґрунти поділяються на сильнокислі (pH = 3...4), кислі (pH= 4...5), слабкокислі (pH = 5...6), нейтральні (pH = 6...7), слабколужні (pH = 7...8), лужні (pH = 8...9) і, нарешті, сильнолужні (pH = 9... 11). Найчастіше рослини страждають від підвищеної кислотності, для усунення якої застосовують вапнування ґрунтів — внесення в них вапняків — карбонатів кальцію або магнію. Якщо ж ґрунти відрізняються підвищеною лужністю (солонцюваті і солончакові ґрунти), то для її усунення застосовують гіпсування — внесення розмеленого гіпсу CaSO4 • 2Н2О. Необхідність вапнування або гіпсування ґрунтів встановлюють із урахуванням водневого показника розчину (сольової витяжки); залежно від величини pH визначають за таблицями і дози речовин, які вносять. 17. Гідроліз солей. Означення. Досвід показує, що розчини середніх солей мають лужну, кислу або нейтральну реакцію, хоча вони і не містять ні водневих, ні гідроксильних іонів. Пояснення цьому факту слід шукати у взаємодії солей з водою. Взаємодія іонів солі з водою, що приводить до утворення слабкого електроліту, називається гідролізом солі. 1. Усі солі, утворені слабкою кислотою і сильною основою, піддаються гідролізу. Вони надають розчину лужної реакції (pH > 7). 2. Солі, утворені сильною кислотою і слабкою основою, також піддаються гідролізу. Вони надають розчину кислої реакції, як це має місце в розчині хлориду амонію NH4Cl. В цьому разі утворюється слабкий електроліт NH4OH. У результаті частина іонів ОН- зв’язується іонами NH+4 , а іони Н+залишаються в надлишку. Отже, внаслідок гідролізу NH4Cl розчин цієї солі набуває кислої реакції (pH > 7). Рівняння гідролізу можна записати так: NH+4 + Н2О ⇆ NH4OH + Н+, або точніше NH+ + Н2O ⇆ NH3 ⇆ Н2O + Н+. 3. Ще легше піддаються гідролізу солі, утворені слабкою кислотою і слабкою основою. Наприклад:CH3COONH4. Іони цієї солі одночасно зв’язують іони Н+ і OН-, зміщуючи рівновагу дисоціації води: СН3СОО- + NH+4 + Н2O ⇆ СН3СООН + NH4OH (NH3 • Н2О). У цьому разі реакція розчину залежить від ступеня дисоціації продуктів гідролізу — кислоти й основи; якщо переважають іони ОН- , вона лужна, а якщо іони Н+ — кисла, якщо ж їх число однакове — нейтральна. Оскільки у прикладі, що розглядається, ступені дисоціації СН3СООН і NH4OH, які утворюються внаслідок гідролізу, приблизно однакові, то розчин солі буде нейтральним. Однак реакція водного розчину карбонату амонію (NН4)2СО3 — також солі слабкої кислоти і слабкої основи — слабколужна: NH+4 + СО2-3 + Н2O ⇆ NH4OH + НСО-3, оскільки ступінь дисоціації NH4OH більший, ніж ступінь дисоціації іона НСО3 . 4. Солі, утворені сильною основою і сильною кислотою, гідролізу не піддаються. Іони таких солей не можуть утворювати з водою слабких електролітів. У цьому випадку солі практично в реакції участі не беруть, і рівновага дисоціації води не порушується, концентрація Н+- і ОН- -іонів залишається такою самою, як і в чистої води, а значить, розчин матиме нейтральну реакцію (pH = 7). Складання рівнянь гідролізу солей. Гідроліз солей, утворених слабкими багатоосновними кислотами і сильними основами, відбувається ступінчасто (відповідно зворотному процесу — ступінчастій дисоціації), і при цьому утворюються кислі солі (точніше, аніони кислих солей). Аналогічно під час гідролізу солей, утворених багатокислотними слабкими основами і сильними кислотами, утворюються основні солі (точніше, катіони основних солей). Гідроліз відбувається в основному за першим ступенем 18. Комплексні сполуки: типи, номенклатура. Комплекси можуть існувати в розчині, кристалі, а інколи і в газовому стані, наприклад [Аl2Сl6]. Заряд комплекса визначають алгебричною сумою зарядів його складових частин. При цьому ступінь окиснення центрального атома приймають за заряд іона. Для лігандів – нейтральних молекул – заряд прирівнюють до нуля, а для аніонних лігандів беруть такий же заряд, як і у відповідного аніона. Комплекс матиме характер катіона або аніона, якщо сумарний заряд координованих груп не дорівнює заряду центрального іона. У цьому разі координаційна сполука має зовнішню сферу, або позасферні іони. Отже, комплексними, або координаційними називають сполуки, кристалічні ґратки яких складаються з поліатомних угруповань (комплексів), здатних існувати самостійно. – У молекулі будь-якої комплексної сполуки один з іонів, найчастіше позитивно заряджений, займає центральне місце і називається комплексо-утворювачем, або центральним іоном. – Атоми, молекули або іони, розташовані безпосередньо навколо комплексоутворювача і зв'язані з ним за рахунок додаткової валентності, називаються лігандами. – Ліганди разом з комплексоутворювачем складають внутрішню сферу комплексу, або внутрішню координаційну сферу, яку при написанні формули координаційної сполуки пишуть у квадратних дужках. – Іони, або нейтральні частини комплексу, що не є лигандами чи комплексоутворювачем і не увійшли у внутрішню координаційну сферу, складають зовнішню сферу комплексів. НОМЕНКЛАТУРА КОМПЛЕКСНИХ СПОЛУК Назви комплексних сполук будують за тими ж принципами, що й назви простих, з урахуванням їхньої хімічної природи. Спочатку називають комплексний катіон (або простий катіон) у називному відмінку, потім – простий аніон (або комплексний аніон). Якщо сполука неелектролітного типу, то її називають одним словом. Називаючи комплексні іони, насамперед вказують ліганди (L) – аніонні, нейтральні та катіонні, а потім центральний атом (М), не розділяючи їх. Формули комплексів записують у зворотному порядку [М(L+)(L0)(L–)]. Назви лігандів одержують сполучну голосну – о, наприклад: Н– – гідридо-, F– – фторо-, О2– – оксо-, О22– – пероксо-, S2– – тіо-, ОН– – гідроксо-, СN– – ціано-, NСО– – ціанато-, SСN– – тіоціанато-, NСS– – ізотіоціанато-, NО2– – нітро-, ОNО– – нітрито-, NН2– – амідо-, NН2– – імідо-, С2О42– – оксалато-, NН2(СН2)2NН2 – етилендіамін-, Н2О – аква-, NН3 – амін-, СО – карбоніл-, NО – нітрозил-, Н+ – гідрогено-, Н3О+ – оксоно-, NН4+ – амоно-, N2Н5+ – гідразино-. Перед лігандами зі складними назвами, які уже містять числівники, для позначення числа лігандів використовують помножуючі префікси біс-, трис-, тетракіс- і т. ін. Назви таких лігандів звичайно беруть у дужки. Якщо ліганд є містком між двома центрами координації, перед його назвою ставлять грецьку літеру μ (мю) через дефіс. Місткові ліганди називають останніми. Щоб уникнути двозначності, складні групи атомів у молекулі відокремлюють різною формою дужок у такому порядку: [{( )}]. 19. Галогени, властивості,сполуки,застосування,отримання. ЙОД ЙОД - І хімічний елемент VII групи періодичної системи Менделєєва, належить до галогенів (у літературі зустрічається також символ J); атомний номер 53, атомна маса 126,9045; кристали чорно-сірого кольору з металевим блиском. Природний йод складається з одного стабільного ізотопу з масовим числом 127. Йод відкрив в 1811 р. французький хімік Б. Куртуа. Нагріваючи матковий розсіл золи морських водоростей з концентрованою сірчаною кислотою, він спостерігав виділення фіолетової пари (звідси назва йод - від грецького iodes, ioeides - схожий кольором на фіалку, фіолетовий), що конденсувався у вигляді темних блискучих пластинчастих кристалів. В 1813 – 1814 рр. французький хімік Ж. Л. Гей-Люссак й англійський хімік Г. Дэві довели елементарну природу йоду. ХЛОР ХЛОР (Cl) - хімічний елемент VII групи періодичної системи Менделєєва, атомний номер 17, атомна маса 35,453; відноситься до галогенів. При нормальних умовах жовто-зелений газ із різким дратівним заходом. Природний хлор складається із двох стабільних ізотопів: 35Cl (75,77%) і 37Cl (24,23%). Xлор отриманий уперше в 1774 р. Шееле взаємодією соляної кислоти з піролюзитом Мn2. Однак, тільки в 1810 М. Дэві встановив, що хлор - елемент і назвав його chlorine (від грецького chloros - жовто-зелений). В 1813 Ж.Л. Гей-Люссак запропонував для цього елемента назву хлор. Xлор зустрічається в природі тільки у вигляді сполук. З киснем хлор утворить оксиди: Cl2O, Cl2, Cl2O6, Cl2O7, Cl2O8, а також гіпохлорити, хлорити, хлорати й перхлорати. Всі кисневі з'єднання хлору утворять вибухонебезпечні суміші з легкоокисними речовинами. Окисиди хлору малостійкі й можуть мимовільно вибухати, гіпохлорити при зберіганні повільно розкладаються, хлорати й перхлорати можуть вибухати під впливом ініціаторів. ФТОР ФТОР (F) - хімічний елемент VII групи періодичної системи Менделєєва, відноситься до галогенів, атомний номер 9, атомна маса 18,998403; при нормальних умовах (0 С; 0,1 Мн/м2, або 1 кгс/см2) - газ блідо-жовтого цвіту з різким заходом. |