Шпора. Экзамен по химии. 1. Основные понятия в химии вещество, молекула, атом. Строение атома. Химический элемент. Изотопы. Атомная единица массы. Число Авогадро. Моль
 Скачать 0.55 Mb. Скачать 0.55 Mb.
|

|
11.Скорость химической реакции. Гомогенные и гетерогенные реакции. Кинетическое уравнение реакции. Период полупревращения. Скорость химической реакции — изменение количества одного из реагирующих веществ за единицу времени в единице реакционного пространства. Является ключевым понятием химической кинетики. Скорость химической реакции — величина всегда положительная, поэтому, если она определяется по исходному веществу (концентрация которого убывает в процессе реакции), то полученное значение умножается на −1. Скорость химической реакции в каждый момент времени пропорциональна концентрациям реагентов, возведенным степени, равные их стехиометрическим коэффициентам. Гомогенные реакции - хим. р-ции, протекающие полностью в одной фазе. Примеры Г. р. в газовой фазе: термич. разложение оксида азота 2N2O5 -> 4NO2 + О 2; хлорирование метана СН 4 + С12 -> СН 3 С1 + НС1; горение этана 2С 2 Н б + 7О 2 -> 4СО 2 + 6Н 2 О; образование озона в земной атмосфере ЗО 2 -> 2О 3. В жидкой фазе гомогенно протекают разнообразные гомологич. р-ции распада молекул на радикалы, нуклеоф. и электроф. замещения, отщепления, перегруппировки, а также цепные р-ции (галоидирование, окисление, полимеризация). Скорость простой Г. р. при постоянном объеме подчиняется действующих масс закону. Г. р. между реагентами, первоначально находящимися в разных фазах, возможны при условии, что диффузия реагентов в ту фазу, где идет р-ция, много быстрее самой р-ции. При относительно медленной диффузии реагентов и быстром хим. взаимод. р-ция протекает на границе раздела фаз. Гетерогенные реакции - химические реакции, протекающие на границе раздела фаз. Для них характерно скачкообразное изменение отдельных параметров. Скорость гетерогенных реакций зависит от площади поверхности соприкосновения реагирующих веществ. Для увеличения скорости реакций твердые вещества измельчают. Примером гетерогенных реакций является горение каменного угля С + О2 ® СО2. Период полупревращения (T1/2) – время по истечении которого остаётся ровно половина от исходного вещества (С=С0/2). Выражение T1/2 для реакции первого порядка: T1/2 для реакции второго порядка, в отличие от первого порядка период обратно пропорционален исходной концентрации реагента. Кинетическое уравнение, выражает зависимость скорости хим. реакции от концентраций компонентов реакционной смеси. Для простой (одностадийной) гомогенной реакции скорость пропорциональна произведению концентраций реагирующих веществ. Кинетическое уравнение реакций первого порядка имеет вид: V=k*C H2S2O3 S(осадок)+SO2(газ)+Н2О (уравнение первого порядка) Кинетическое уравнение реакций второго порядка имеет вид: V=kCA*CB В частном случае когда реагируют одинаковые молекулы, уравнение принимает вид: V=kc2 2HI(газ) H2(газ)+I2(газ) -dc/dt=kc2, разделяем переменные и при начальных условиях, при t=0, С=С0 получаем: 1/С+1/С0+kt Последнее уравнение и является решением 12.Влияние температуры на скорость реакции. Правило Вант-Гоффа. Энергия активации. Уравнение Аррениуса. Эндотермические и экзотермические реакции Константа скорости реакции есть функция от температуры; повышение температуры увеличивает константу скорости. Правило Вант-Гоффа- скорость большинства реакций увеличивается в 2-4 раза при повышении t на каждые 10градусов: v2/v1 = γ(T2-T1)/10 ,где v1 и v2 – скорости реакции при температурах Т1 и Т2; величина g называется температурным коэффициентом реакции, показывает во сколько раз увеличится скорость реакции при увеличении t на 10градусов. Если за скоростью реакции следить при постоянной исходной конц реагентов,то в этом случае уравнение также справедливо для отношения const V: k2/k1 = γ(T2 - T1)/10 ,где k1,k2-const скорости реакции при T1,2.Более строгую зависимость скорости реакции от t на основании экспериментальных данных вывел Аррениус: где A-предэкспоненциальныи множитель (размерность совпадает с размерностью к), Еа-энергия активации, обычно принимающая положит. значения, Т-абс. т-ра, k-постоянная Больцмана. Энергия активации- минимальная энергия, которой должны обладать молекулы, чтобы взаимодействие между ними стало возможным. За очень малые промежутки времени реакции протекают через образование промежуточных состояний системы, называется промежуточным(активирующим) комплексом. В активирующем комплексе старые связи ослабевают ,но не разрываются. Новые связи еще не образовались ,но уже наметились. При распаде комплекса выделяется энергия ,равная Еа обратной реакции. Если при распаде переходного комплекса Е выделилось больше, чем было затрачено на активацию системы, то химический процесс в целом протекает с выделением теплоты- экзотермическая реакция.( 2H2 + O2 → 2 H2O).Если выделилось меньше, чем Еа прямой реакции, то химический процесс протекает с поглощением теплоты- эндотермическая реакция(CaCO3=CaO+CO2). Таким образом, Q р-ции=Еа(обратной)-Еа(прямой) реакции. Чаще Q реакции выражается через изменение энтальпии реакции(ΔH), которую находят как: ΔHр= Hпрод – Hисх. Для экзотермических р-ций(Q>0) ΔHр<0, для эндотермических наоборот. Q=- ΔHр 13. Кинетика обратимых реакций. Химическое равновесие, выражение для константы равновесия, сдвиг химического равновесия. Принцип ле Шателье Химические реакции часто являются двусторонними (обратимыми), т.е. могут протекать при данных условиях в двух противоположных направлениях. двусторонняя реакция обратима в состоянии химического равновесия. А + В <––> D + E Скорость уменьшения концентрации вещества А при протекании прямой реакции определяется уравнением , а скорость возрастания концентрации вещества А в результате протекания обратной реакции – уравнение Общая скорость двусторонней реакции в любой момент времени равна разности скоростей прямой и обратной реакции: Химическое равновесие. Принцип Ле Шателье. С помощью кинетических уравнений прямой и обратной реакций можно вывести закон действующих масс для химического равновесия. Пусть происходит обратимая реакция: Здесь а, b, c, d – коэффициенты перед веществами в химических уравнениях прямой и обратной реакций. В этом случае можно записать кинетические уравнения: vпр = k1[A]a[B]b; vобр = k2[C]c[D]d При наступлении равновесия скорости прямой и обратной реакции становятся равны (vпр = vобр) и можно записать: k1[A]a[B]b = k2[C]c[D]d Из этого соотношения можно получить константу равновесия Кр, которая равна отношению констант скорости прямой и обратной реакций: В предыдущем уравнении достаточно перенести в левую часть k2 а из полученного уравнения уже легко получается выражение для константы равновесия Кр: Это не что иное, как математическая запись закона действующих масс для химического равновесия. Например, для рассмотренной выше реакции 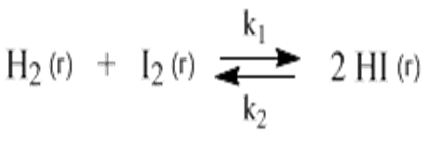 Численное значение Кр характеризует положение равновесия при данной температуре и не меняется с изменением концентраций реагирующих веществ. vпр = k1[CO]; vобр = k2[CO2] В этом случае говорят, что наступило химическое равновесие. Количество содержащихся в равновесной системе веществ H2, I2 и HI теперь не меняется со временем, если нет внешних воздействий на систему. Химическим равновесием называется такое состояние химической системы, при котором количества исходных веществ и продуктов не меняются со временем. принцип Ле Шателье: Если на равновесную систему воздействовать извне, изменяя какой-нибудь из факторов, определяющих положение равновесия, то в системе усилится то направление процесса, которое ослабляет это воздействие. для смещения равновесия вправо нужно: 1 повышать давление, 2 понижать температуру добавление катализатора не может изменить положение равновесия, но способствует более быстрому его достижению. 14.Гомогенный и гетерогенный катализ, цели применения катализаторов. Ферментативный катализ и его особенности. Катализ- химический процесс, протекающий в присутствии катализаторов. Катализаторы-вещества, резко изменяющие скорость реакции, но при этом в ней не расходуются. Катализаторы позволяют:-1)изменить скорость реакции (как замедлить, так и увеличить)-если к раствору Н2О2 добавить небольшое количество Н2SO4конц,то скорость разложения Н2О2 замедлится почти до 0.Напротив, если +MnO2, то будет наблюдаться бурное разложение: 2H2O2=2H2O + O2. Катализаторы, замедляющие скорость реакции-ингибиторы.-2)изменить направление процесса:C2H5OH=C2H4+H2O(при условии Al2O3,t=300); C2H5OH=CH3-COH+H2(при условии Cu,t=300).-3)снизить энергозатраты: при введении катализатора снижается Ea реакции и тем самым уменьшается Е барьер(реакция будет протекать при более низкой t) Различают гомогенный и гетерогенный катализ .При гомогенном- реагирующие вещества и катализатор находятся в одной фазе. При гетерогенном- реакционные смеси и катализатор- в разных фазах. Ферментативный катализ Практически все биохим. реакции, протекающие в живых организмах, идут под действием биологических катализаторов - ферментов .Ферменты(энзимы)-белковые молекулы, которые катализируют химические реакции в живых системах. В ферментах выделяют активные центры- определенные участки белковых молекул, захватывающих субстрат(различные вещества), причем разложенные направляются по строго определенному пути, и аллостерические центры - участки ферментов, которые распознают субстрат и способствуют его размещению в активном центре. Специфические свойства ферментов 1.Размеры-молекулярная m=10^5-10^7 2.реакции протекающие под действием ферментов ,относят к спец.классу ультрамикрогетерогенных(т.к. размеры молекул ферментов сопоставляются с размерами коллоидных частиц) 3.Чрезвычайно высокая каталитическая активность.(T1/2 мочевины составляет 10^9сек. Ферментативный катализ имеет две характерные особенности: 1.Высокая специфичность. Действие ферментов носит строго ориентированный характер. Так уреаза разрушает мочевину, алкогольдегидрогеназа- группу спиртов. Амилаза катализирует процесс расщепления крахмала, представляющего собой цепь одинаковых глюкозных звеньев, но не катализирует гидролиз сахарозы. 2.Высокая активность, на несколько порядков превышает активность неорганических катализаторов, что объясняется значительным снижением энергии активации процесса ферментами.(константы скорости гидролиза мочевины в присутствии кислоты и уреазы различаются на 13 порядков, составляя 7,4*10^-7 и 5*10^6 c^-1. 15. Тепловой эффект реакции. Первый закон термодинамики. Понятие энтальпии. Закон Гесса. Калорийность пищи. Тепловой эффект реакции – количество поглощённой или выделившейся в ходе реакции теплоты. Равен разности между Еа обратимой и прямой реакции. Первый закон термодинамики – закон сохранения и превращения энергии (физик Мейер). Энергия не исчезает и не возникает из ничего., а только превращается из одного вида в другой в строго эквивалентных соотношениях. Если система в замкнутом объеме атомов, то поглощенная энергия идёт на изменение внутренней энергии. Q = ∆ U = U2 – U1 Если система находится под постоянным давлением, то поглощаемая теплота идёт на увеличение внутренней энергии и на совершение работы против внешних сил. Qp = ∆U + A Сумма u + pv называют энтальпией и обозначают Н. Энтальпия – термодинамическая функция состояния системы, зависит только от начального и конечного состояния системы, но не зависит от пути перехода. Тепловой эффект реакции для изобарных процессов = изменению энтальпии. Термохимическое уравнение – уравнение в котором указан тепловой эффект реакции. Зная стандартные энтальпии образования по закону Гесса можно найти тепловой эффект реакции. ∆Н = ∑ Энтальпия реакции равна разности сумм энтальпии образования всех продуктов реакции и сумме энтальпии образования всех исходных веществ. - стехеометрический коэффициент и энтальпия образования - продукта, - исходных веществ. Al2O3 + 3SO2 => Al2(SO4)3 Калорийность пищи: При окислении продуктов питания в организме выделяется теплота. Калорийность пищи – энергия, выделяемая при полном окислении 1г питательных веществ. Измеряется энергетическая ценность продуктов в кКалл/г 1калл = 4,19 Дж Калл – количество теплоты, которое надо сообщить 1г воды чтобы нагреть его на 1С (масло сливы – 7,26, жир – 9) 16.Энтропия. Второй закон термодинамики. (постулат Планта): Энергия Гиббса. Все химические процессы можно разделить на 2 типа: Протекающие самопроизвольно (окисление металлов) Не протекающие самопроизвольно Самопроизвольным, или спонтанным, является процесс, который совершается в системе без затраты работы извне и который уменьшает работоспособность системы после своего завершения. Критерии самопроизвольной протекании реакции является: Энтальпический фактор – как правило самопроизвольно протекают процессы, идущие с выделением тепла. Энтропийный фактор – в самопроизвольных процессах неупорядоченность системы возрастает. Энтропия – термодинамическая функция, характеризующая меру неупорядоченности системы. Энтропия может быть охарактеризована числом микросостояний: Числом мгновенных координат и скоростей молекул, образующих химическую систему, так как это число огромно, то для количественной характеристики берется логарифм от числа микросостояний. В этом случае энтропия равна:  [ [ ] (1) ] (1)Где R – газовая постоянная равна 8,31 Дж/моль∙К; W – число микросостояний. В отличии от внутренней энергии и энтальпии, которые измерить абсолютно невозможно, энтропию можно измерить непосредственно, что вытекает из 3 закона термодинамики (постулат Планта): Энтропия индивидуального кристаллического вещества при абсолютном нуле равна нулю: S0=0. Постулат планка справедлив только для индивидуальных веществ, кристаллы которых идеально построены (в кристаллической решетке все узлы заняты молекулами или атомами, правильно чередующимися и закономерно ориентированными). Такие кристаллы называются идеальными твердыми телами. Max Энтропией обладают газы. При усложнении молекул(повышении числа атомов) энтропия также возрастает. Энтропия вычисленная при стандартных условиях (T=298 К, 25 ̊С, р=1 атмосфере) называется стандартной энтропией S0 (табличные данные). Изменения энтропии в ходе реакции находится по аналогии с изменениями энтальпии:  (2) (2)Энтропия простых веществ ≠0 Второй закон термодинамики. Второй закон термодинамики определяет направленность и пределы протекания самопроизвольных процессов, в том числе и биохимических. В изолированных системах самопроизвольно идут только те процессы, которые сопровождаются увеличением энтропии (ΔS>0). Таким образом, при протекании химических реакций возможно 2 противоположные тенденции: Образование более прочных упорядоченных систем, сопровождающихся снижением энергии (Δh<0)$ Стремление к беспорядку количественно характеризуется возрастанием энтропии (ΔS>0). Характер протекания химических процессов определяется соотношением этих 2-х факторов – энтальпийного и энтропийного. Для неизолированных систем нужно учитывать не только изменения энтропии, но и изменение энергии. Все процессы, при которых энергия в системе уменьшается, а энтропия возрастает, протекает самопроизвольно. Самопроизвольность других процессов зависит от того, какая из этих 2-х тенденций – энергетическая или энтропийная – скажется более эффективной, какая из этих противоборствующих тенденций получит перевес над другой. Энергия Гиббса. Биохимические реакции обычно происходят при изобарно-изотермических условиях. В этих случаях энергетическое состояние системы характеризуется энтальпией, а мерой неупорядоченности системы будет произведение ее энтропии и температуры. Функцией, учитывающей обе эти характеристики и противоположность в тенденции их изменения при самопроизвольных процессах, является энергия Гиббса. Свободная энергия Гиббса (G) – термодинамическая функция объединяющая энтальпийный и энтропийный факторы и являющиеся критерием самопроизвольного протекания химический реакции.  (3) (3)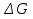 – изменение энергии Гиббса в ходе химической реакции; – изменение энергии Гиббса в ходе химической реакции;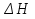 и и 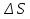 - соответственно изменения энтальпии и энтропии; - соответственно изменения энтальпии и энтропии;T – температура, при которой протекает реакция. Как следует из формулы (3) энергия Гиббса может быть сосчитана изменением энтальпии и энтропии с учетом Исходя из полученного значения, можно судить о возможности протекания данной химической реакции. Возможны следующие 3 случая: <0 – самопроизвольное протекание химической реакции возможно;>0 – реакция протекать самопроизвольно не может;=0 – химическая система находится в равновесие (обратима).Энергия Гиббса. Образование вещества. - изменение энергии Гиббса при образовании 1 моль вещества из простых веществ. Если изменение энергии Гиббса рассматривать при стандартных условиях, то говорят о стандартном изменении энергии Гиббса. 0 образование простых веществ принимается равным 0.0 – табличные данныеЭнергия Гиббса – термодинамическая функция зависит только отначального и конечного состояния системы поэтому изменение энергии Гиббса в ходе реакции вычисляется по полной аналогии с изменениями энтальпии и энтропии. 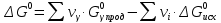 (4) (4)При достижении равновесия химической системы константа равновесия связана со стандартными изменениями энергии Гиббса соотношение: 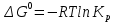 (5) (5)Где 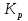 - константа равновесия при температуре Т - константа равновесия при температуре ТИсходя из изменения энергии Гиббса в ходе реакции можно рассчитать константу равновесия при произвольной температуре Т: 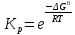 (6) (6)Также может быть решена обратная задача: по экспериментальному определению рассчитывает из уравнения (5) изменения стандартной энергии Гиббса. |