Окисление. Федеральное агентство по образованию
 Скачать 1.34 Mb. Скачать 1.34 Mb.
|

|
11. ОКИСЛЕНИЕ КАРБОНОВЫХ КИСЛОТ При действии пероксида водорода в присутствии кислотного катализатора на карбоновые кислоты образуются перкислоты (надкислоты):  Наиболее распространенным катализатором в случае алифатических карбоновых кислот является концентрированная H2SO4. Реакция обратима, и равновесие можно сместить вправо, удаляя воду или применяя избыток реагента. Для субстратов с ароматическими группами R наилучшим катализатором является метансульфокислота, которая используется и как растворитель. Карбоновые кислоты подвергаются окислительному декарбоксилированию под действием тетраацетата свинца и в качестве продуктов в зависимости от условий получаются алканы, алкены или эфиры уксусной кислоты:  Для данной реакции предполагается следующий механизм: 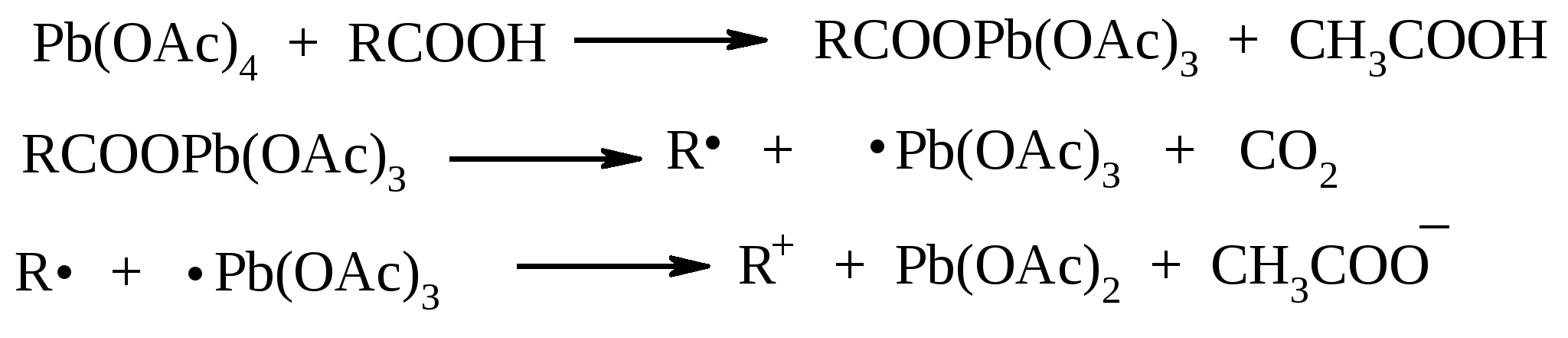 Алканы образуются за счет отрыва атома водорода от молекул растворителя радикалом R, а алкен и сложный эфир из карбокатиона соответственно за счет отщепления протона или захвата ацетат-иона. Введение в реакционную смесь галогенид-иона практически нацело подавляет оба процесса и приводит к образованию алкилгалогенидов. 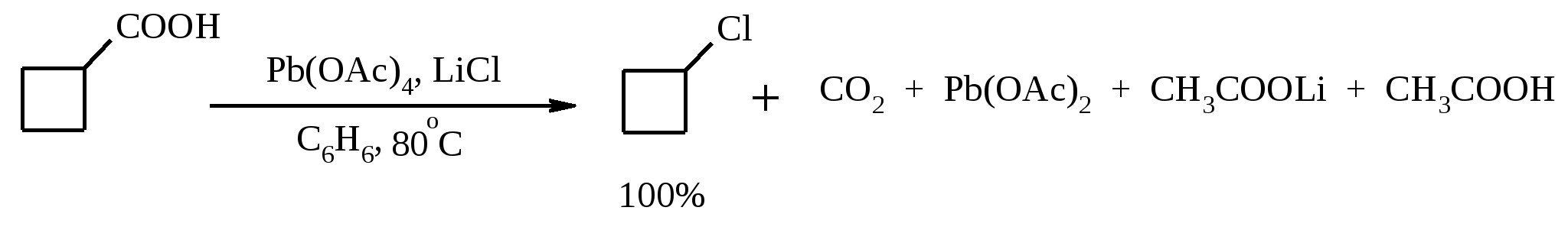 В присутствии иода и тетраацетата свинца карбоновые кислоты превращаются в соответствующие иодиды: 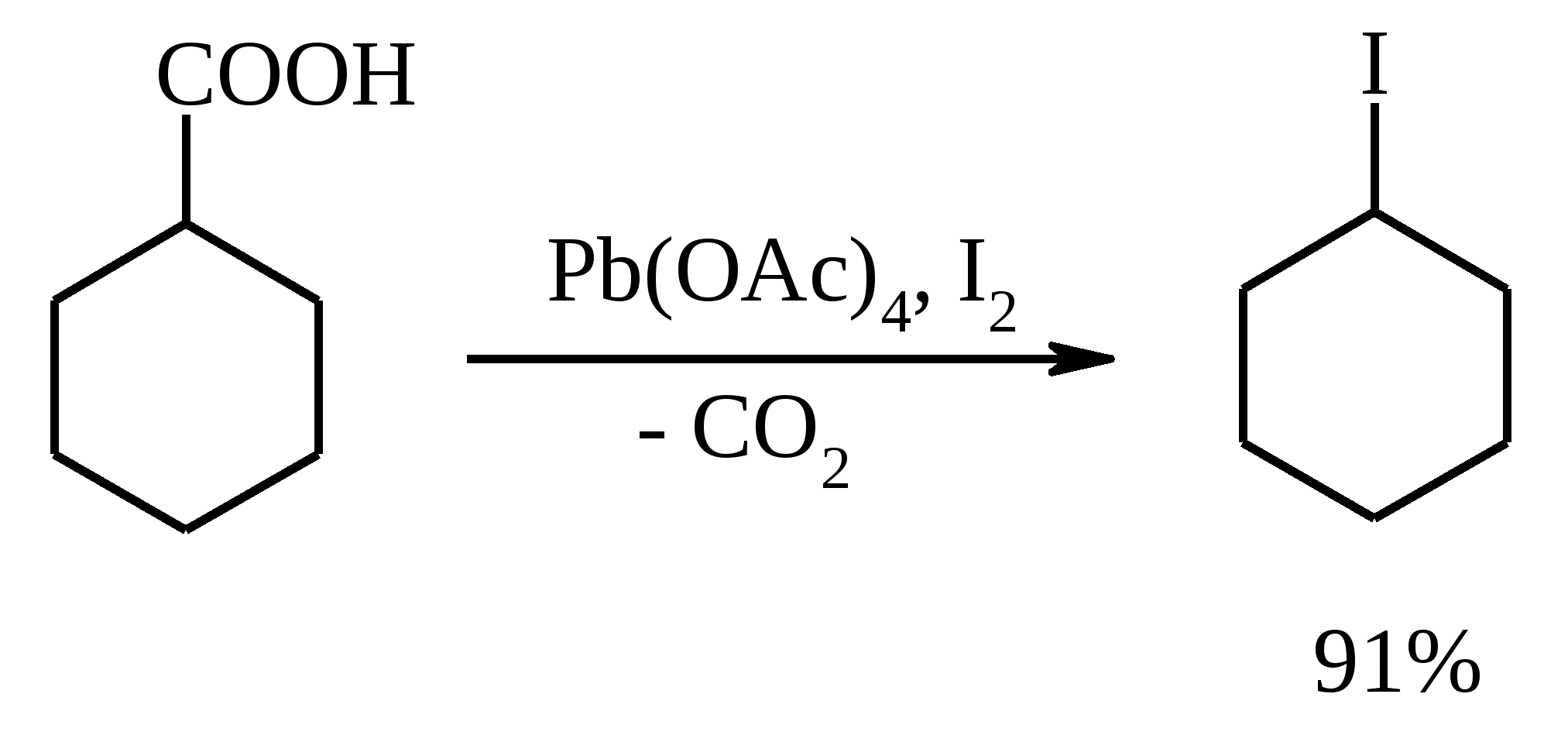 Из 1,2-дикарбоновых кислот под действием тетраацетата свинца образуются алкены: 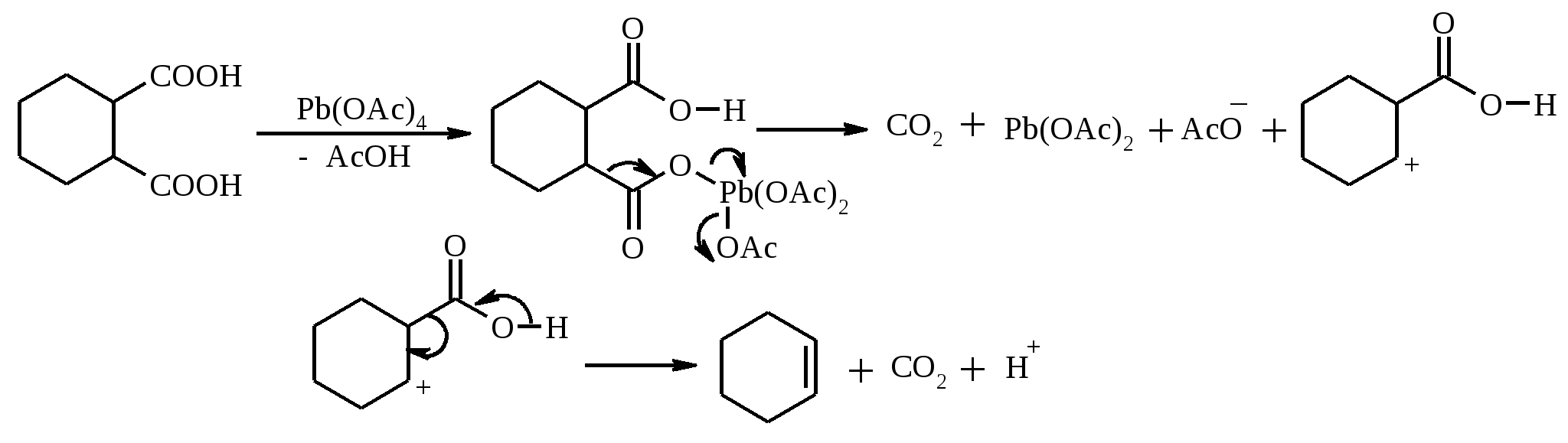 Другим примером окислительного декарбоксилирования может служить окисление серебряных солей карбоновых кислот бромом в CCl4 с образованием алкилбромидов (реакция Хунсдиккера – Бородина): 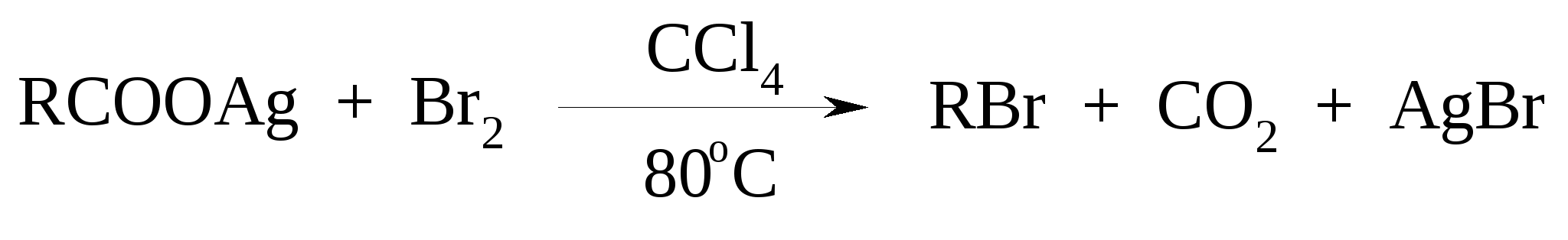 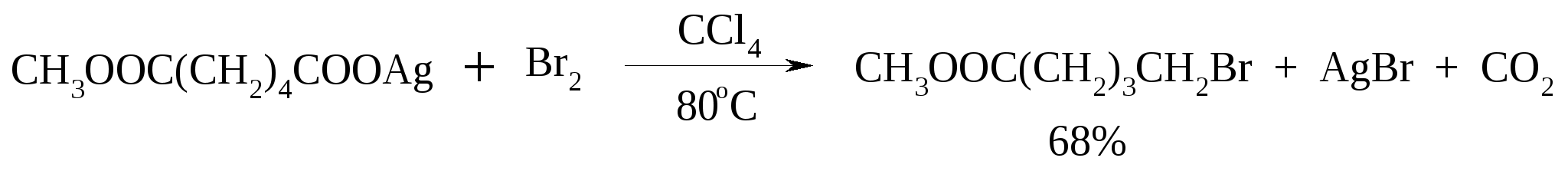 Для успешного проведения реакции требуется применять тщательно высушенные серебряные соли карбоновых кислот, и выход алкилбромида колеблется в широких пределах в зависимости от степени очистки и обезвоживания соли. Этого недостатка лишена модификация с использованием ртутных солей, причем ртутную соль не выделяют индивидуально, а смесь карбоновой кислоты, оксида ртути (II) и брома нагревают в индифферентном растворителе. Этот метод приводит, как правило, к более высоким и воспроизводимым выходам. 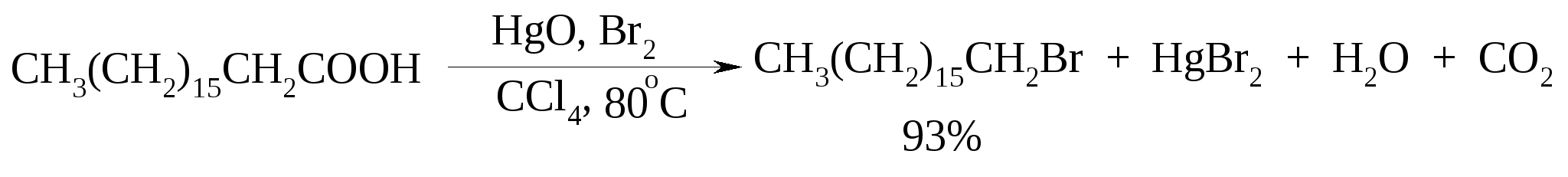 Для реакции Хунсдиккера – Бородина установлен цепной радикальный механизм. Образующийся в первой стадии ацилгипобромит подвергается гомолитическому расщеплению с образованием карбоксильного радикала и атома брома. Карбоксильный радикал теряет CO2 и превращается в алкильный радикал, который затем регенерирует цепь, отщепляя атом брома от ацилгипобромита. 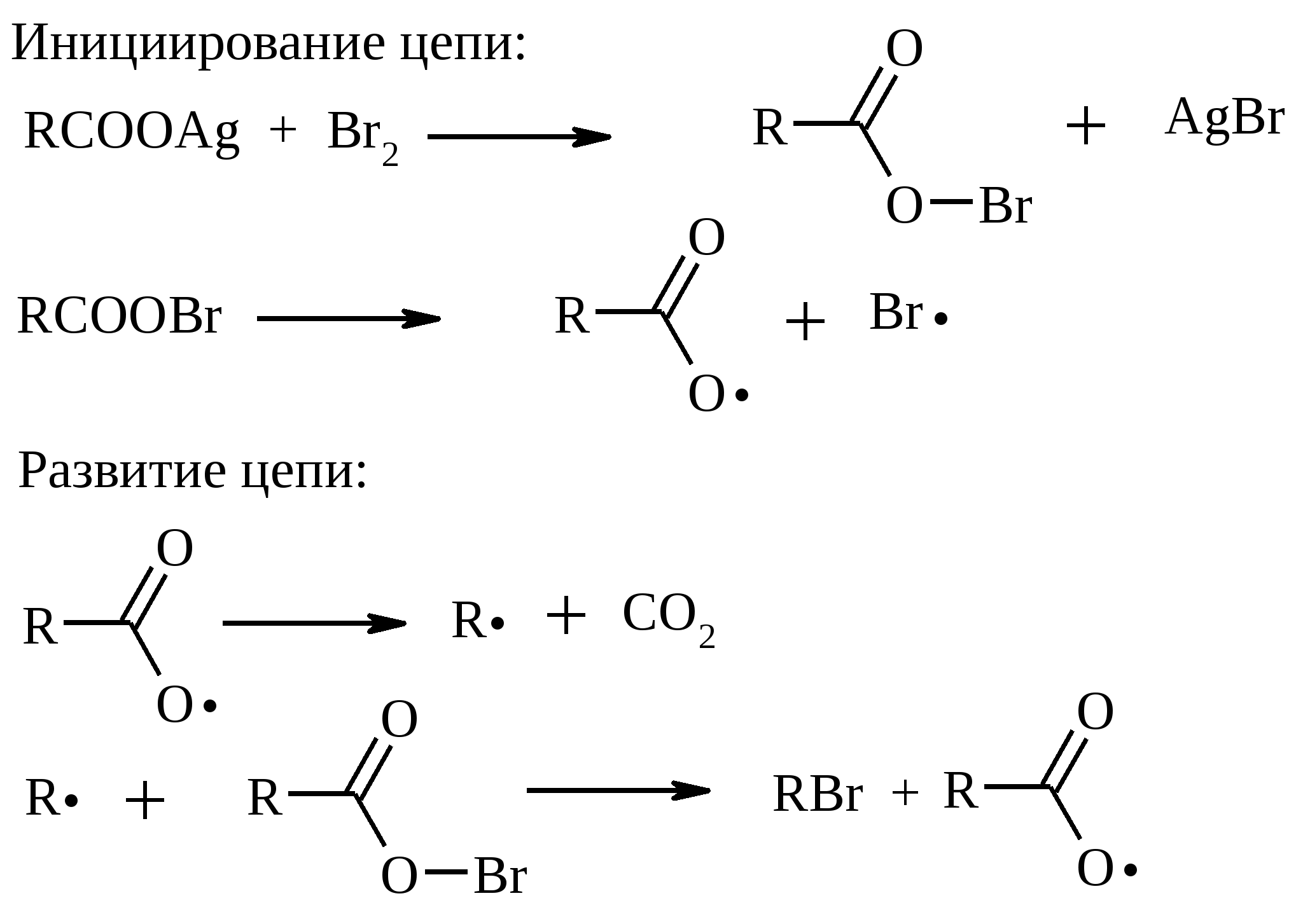 -Гидрокси- и -кетокислоты не расщепляются под действием HIO4, но эта реакция идет с тетраацетатом свинца, H2O2 в щелочной среде и другими реагентами. Такие реакции представляют собой окислительное декарбоксилирование. Из -гидроксикислот получаются альдегиды или кетоны, а из -кетокислот – карбоновые кислоты: 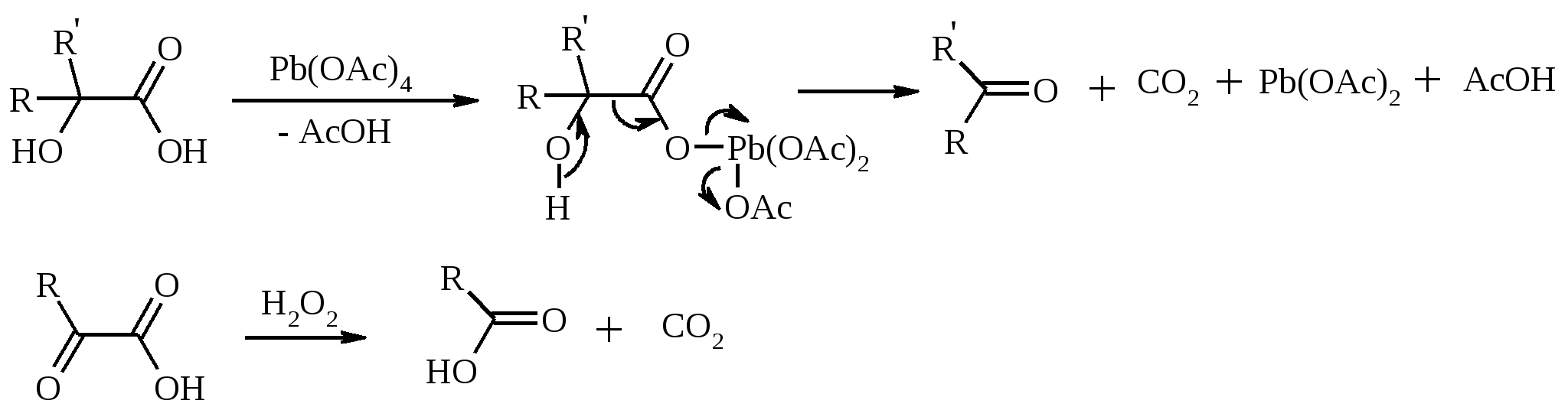 В то же время при использовании пероксида водорода в присутствии Fe(III) удается окислить гидроксильную группу в -гидроксикислотах в карбонильную с сохранением карбоксильной: 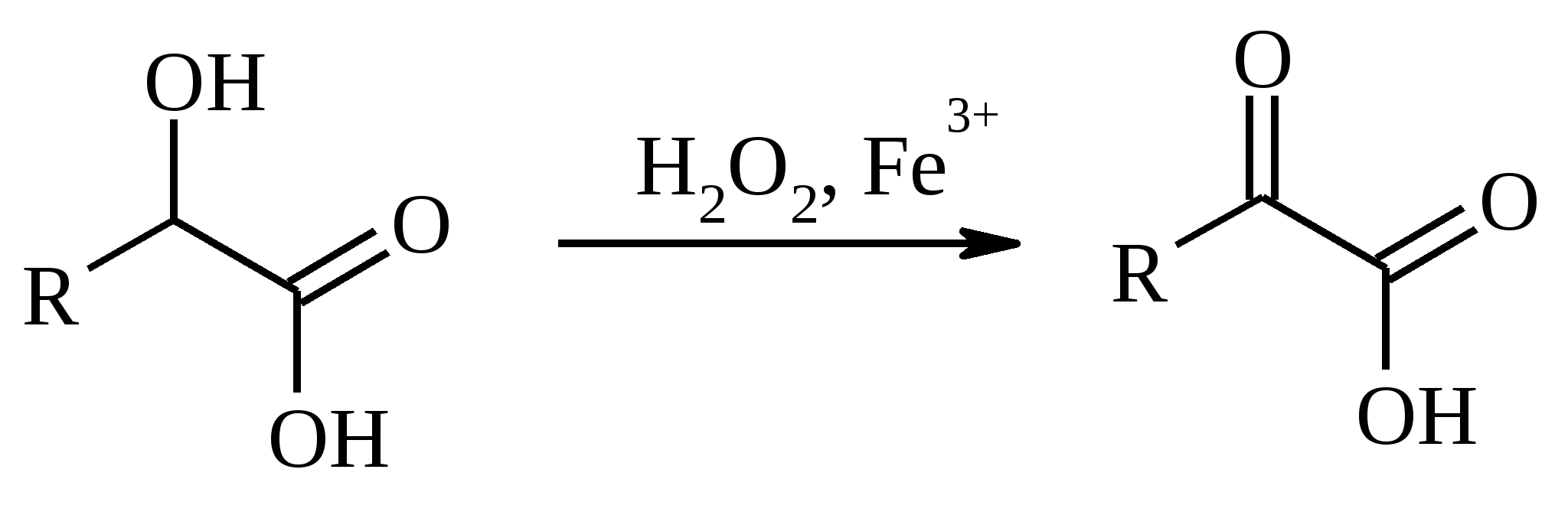 Окислить алифатические карбоновые кислоты в α-гидроксикислоты удается в том случае, если кислота содержит третичный атом углерода в α-положении к карбоксильной группе. В качестве окислителя применяют щелочной раствор KMnO4. 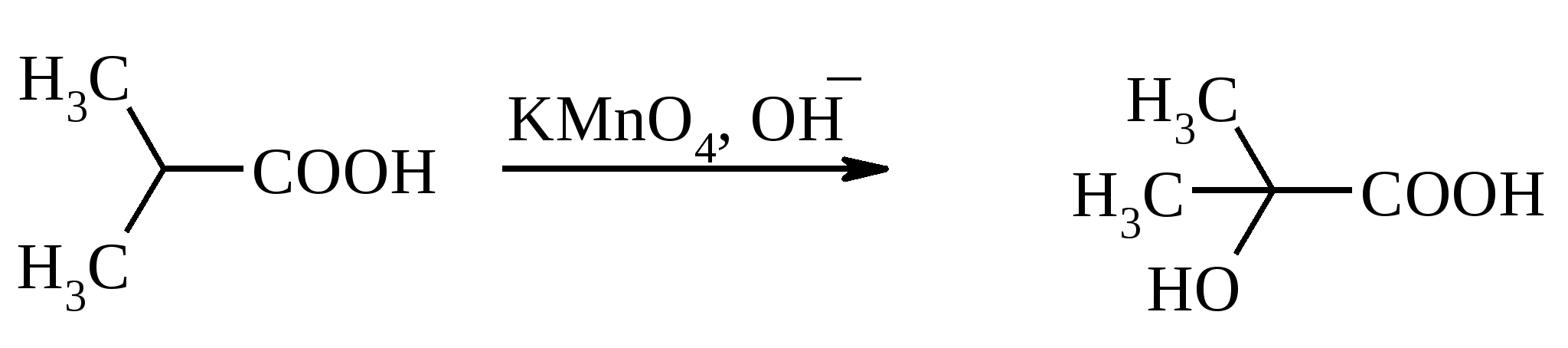 12. ОКИСЛЕНИЕ ПРОСТЫХ ЭФИРОВ Простые эфиры проявляют повышенную склонность к аутоокислению в присутствии кислорода с образованием гидропероксидов. Процесс протекает по цепному радикальному механизму. Эффективным катализатором аутоокисления может служить любой источник свободных радикалов. Аутоокисление простых эфиров представляет большую потенциальную опасность при работе с эфирами в качестве растворителей, поскольку гидропероксиды, накапливающиеся в остатке при перегонке, могут детонировать при слабом перегреве.  Поэтому гидропероксиды должны быть тщательно удалены до перегонки с помощью восстановителей – солей Fe(II) или Sn(II). Простые эфиры, имеющие по крайней мере одну первичную алкильную группу, окисляются в соответствующие сложные эфиры с высокими выходами под действием RuO4, а также при действии CrO3 в серной кислоте, перманганата бензилтриэтиламмония и др.:  Простые эфиры енолов окисляются до сложных эфиров под действием хлорхромата пиридиния:  Под действием 1-хлорбензотриазола простые эфиры подвергаются окислительному расщеплению в альдегиды: 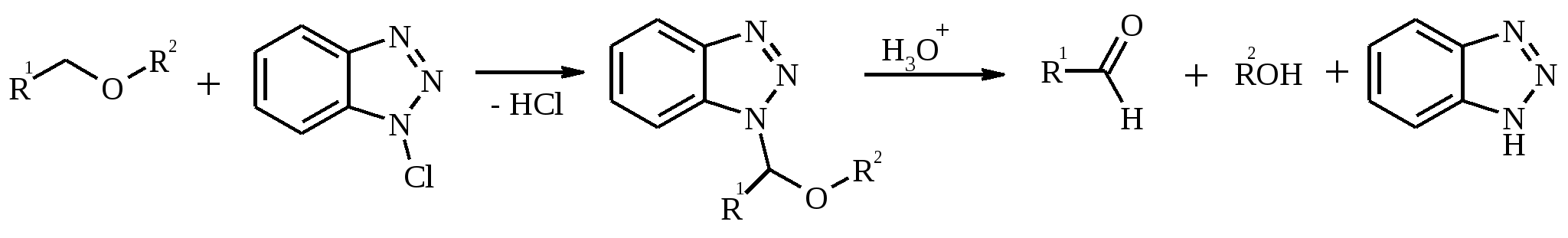 13. ОКИСЛЕНИЕ ЭПОКСИДОВ HIO4 расщепляет эпоксиды в альдегиды и кетоны:  При окислении эпоксидов диметилсульфоксидом образуются -гидроксикетоны или -гидроксиальдегиды:  Сильные окислители превращают эпоксиды в кетоны и (или) карбоновые кислоты: 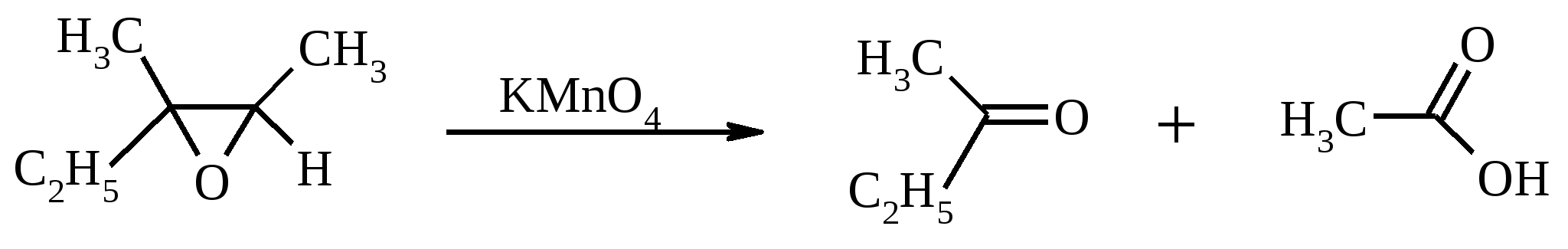 14. ОКИСЛЕНИЕ СЕРОСОДЕРЖАЩИХ СОЕДИНЕНИЙ 14.1. ОКИСЛЕНИЕ ТИОЛОВ Под действием I2, Br2, H2O2, Pb(OAc)4, MnO2 тиолы окисляются до дисульфидов: 2RSH + I2 R-S-S-R Перкислоты окисляют тиолы до сульфиновых кислот:  Сильные окислители – HNO3 и KMnO4 – окисляют тиолы до сульфоновых кислот:  14.2. ОКИСЛЕНИЕ СУЛЬФИДОВ Под действием метапериодата натрия NaIO4, м-хлорнадбензойной кислоты, трет-бутилгипохлорита сульфиды окисляются в сульфоксиды. Наиболее часто применяется 0.5 М водный раствор NaIO4. Этот реагент селективно окисляет сульфиды до сульфоксидов практически без примеси сульфонов, если окисление проводить при 0С в бинарной системе вода – органический растворитель (метанол, диоксан, ацетонитрил):  Механизм окисления аналогичен механизму расщепления 1,2-гликолей и включает циклический интермедиат:  Превращение сульфидов в сульфоксиды под действием трет-бутилгипохлорита можно проиллюстрировать следующим примером:  Окисление сульфидов до сульфонов осуществляется под действием более сильных окислителей (KMnO4, HNO3) или в более жестких условиях при 90-100С с помощью избытка пероксида водорода или трет-бутилгидропероксида в уксусной кислоте:  Диоксираны селективно окисляют сульфиды до соответствующих сульфоксидов (при эквимольном соотношении реагентов) или до сульфонов (при избытке диоксирана). 15. ОКИСЛЕНИЕ АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИЙ 15.1. ОКИСЛЕНИЕ АМИНОВ Все амины сравнительно легко окисляются из-за своей основной природы. Легче всего окисляются до N-оксидов третичные амины. В качестве окислителей используют 30%-ный раствор H2O2 в воде, перкислоты в апротонной среде, диоксираны:   При окислении вторичных аминов вслед за образованием N-окиси происходит миграция протона с образованием N,N-диалкилгидроксиламина:  Первичные амины окисляются намного сложнее, поскольку образующееся производное гидроксиламина может далее окисляться до нитрозосоединений:  При наличии атома водорода при -углеродном атоме нитрозосоединение изомеризуется в оксим:   В более жестких условиях первичные амины окисляются до нитросоединений:  Окисление первичных аминов диметилдиоксираном протекает быстро (от нескольких минут до нескольких часов) в мягких условиях и приводит к соответствующим нитросоединениям с высоким выходом: 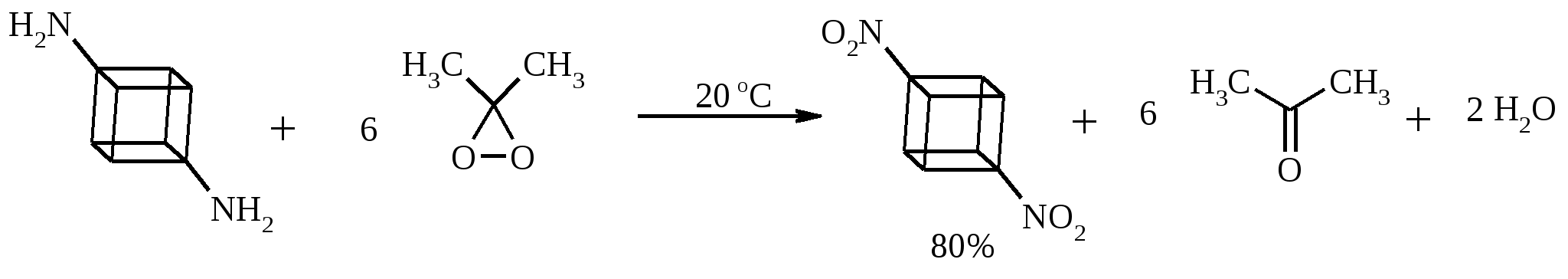 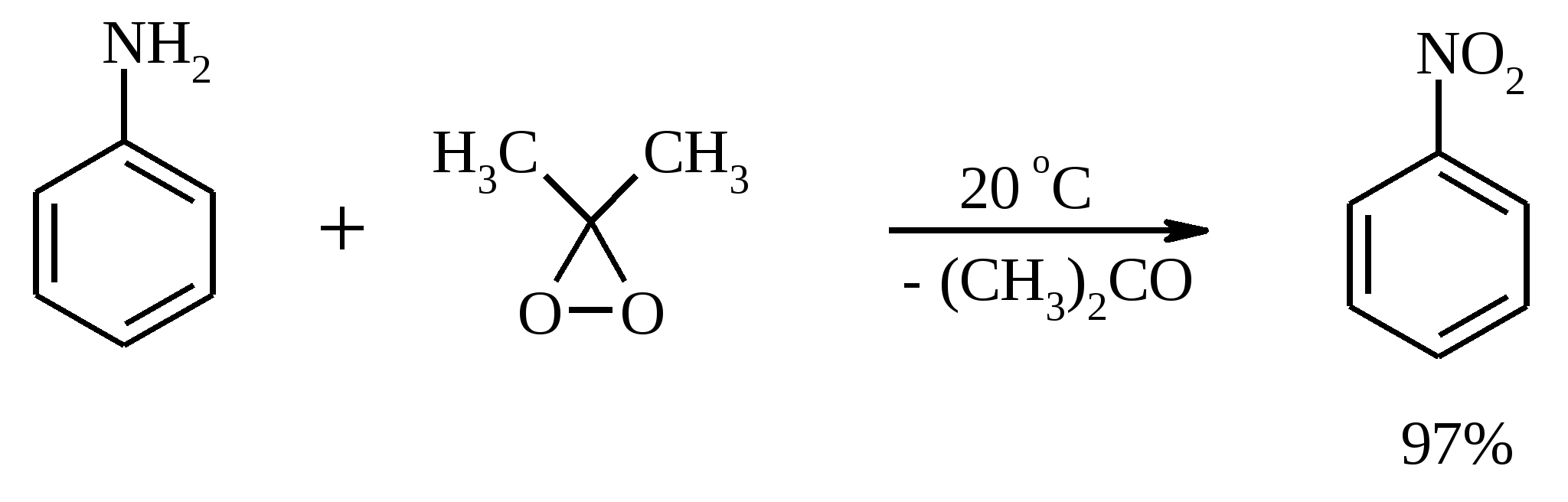 Первичные амины, в которых аминогруппа соединена с третичным атомом углерода, с высоким выходом окисляются в нитросоединения перманганатом калия. Первичные амины, содержащие первичные, вторичные или третичные алкильные радикалы окисляются до нитросоединений сухим озоном, а также различными перкислотами. Первичные, вторичные и третичные алифатические амины расщепляются, давая альдегиды, кетоны или карбоновые кислоты, под действием бромной воды, N-бромсукцинимида, нейтрального раствора KMnO4, нитробензола (для бензиламинов), PdCl2, AuCl3, водного раствора NaOCl в условиях межфазного катализа:  Первичные алифатические амины окисляются в альдегиды или кетоны при взаимодействии с Ag(II), полученным insitu обработкой нитрата серебра персульфатом натрия. Амин сначала дегидрируется до имина, который далее подвергается гидролизу:  Соли бензиламинов дают бензальдегиды или арилкетоны при нагревании в ДМСО:  Дегидрирование первичной аминогруппы, соединенной с первичным атомом углерода, приводит к образованию нитрилов. Реакция осуществлена под действием ряда реагентов: IF5, Pb(OAc)4, CuCl – O2 – пиридин, NBS – (С2H5)3N и др.  Вторичные амины в этих условиях, а также под действием палладиевой черни часто дегидрируются до иминов. Далее имин взаимодействует с молекулой исходного или иного амина, давая аминаль, который теряет аммиак или RNH2, и в результате получается первичный или третичный амин. Ароматические первичные амины под действием MnO2, Pb(OAc)4, Ba(MnO4)2, O2 в присутствии основания окисляются до азосоединений:  15.2. ОКИСЛЕНИЕ ГИДРАЗИНОВ, ГИДРАЗОНОВ, ОКСИМОВ, ГИДРОКСИЛАМИНОВ И АЗОБЕНЗОЛОВ N,N’-Диарилгидразины (гидразосоединения) окисляются в азосоединения при действии ряда окислителей, включая NaOBr, HgO, K3[Fe(CN)6] в условиях межфазного катализа, MnO2 (этот реагент приводит к цис-азобензолам), CuCl2, а также кислород воздуха в присутствии NaOH. Реакция применима также и к N,N’-диалкил- и N,N’-диацилгидразинам. Гидразины (как алкильные, так и арильные), монозамещенные только с одной стороны, также дают азосоединения, однако последние неустойчивы и разлагаются на азот и углеводород: При окислении гидразонов HgO, Ag2O образуются диазосоединения:  Гидразоны ароматических альдегидов взаимодействуют с HgO в таких растворителях, как диглим или этанол, давая нитрилы:  Окисление дигидразонов 1,2-дикарбонильных соединений является одним из лучших современных методов синтеза замещенных ацетиленов. В качестве окислителей используют HgO, Pb(OAc)4, O2 в присутствии СuCl и др.: 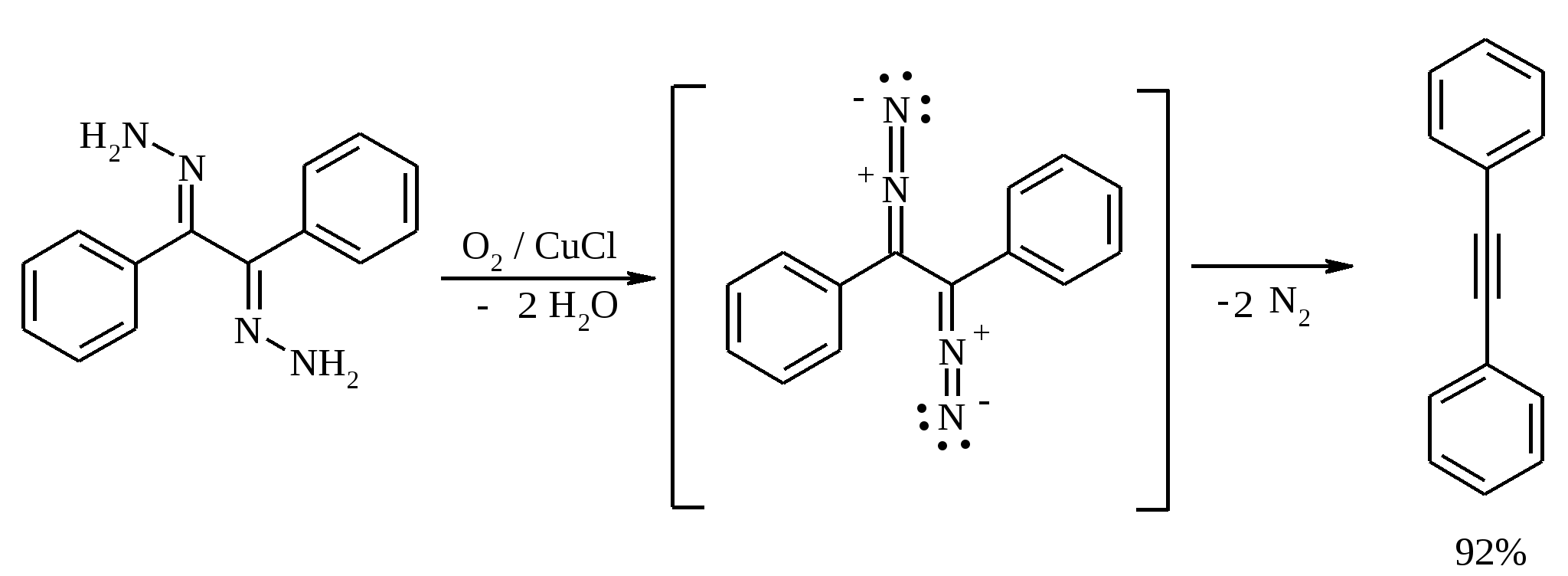 Ароматические гидроксиламины легко окисляются в нитрозосоединения чаще всего под действием Cr(VI) в кислой среде: Окисление оксимов кетонов с помощью трифторперуксусной кислоты в ацетонитриле приводит к нитросоединениям. Первоначально при окислении оксимов образуется аци-форма нитросоединения, которая в кислой среде изомеризуется в нитросоединение:  При окислении кетоксимов диметилдиоксираном также с высоким выходом образуются соответствующие кетоны: 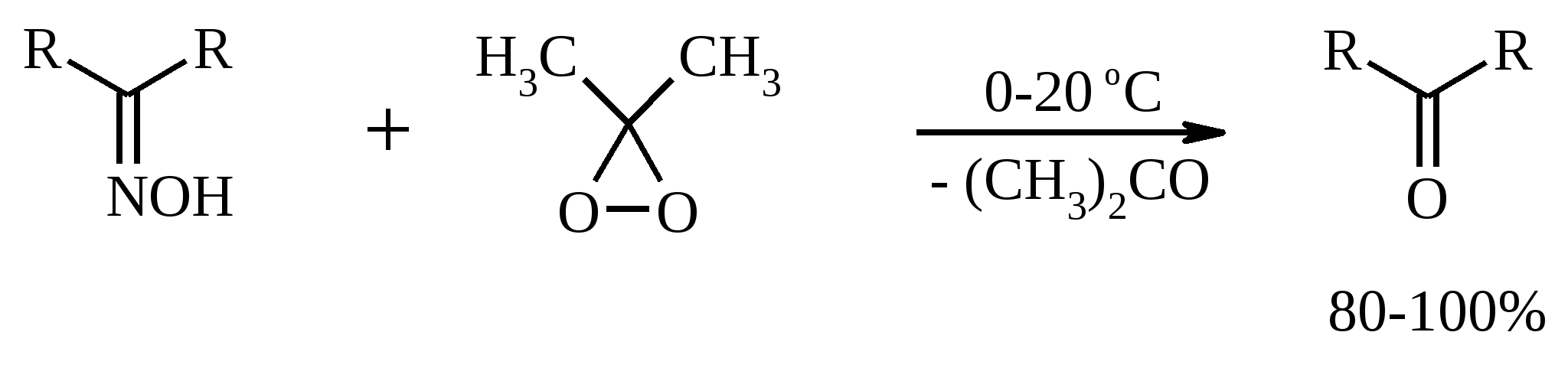 При действии N-бромсукцинимида на оксимы образуются геминальные бромнитрозосоединения (реакция Иффланда), которые далее могут быть окислены в бромнитросоединения под действием водного раствора гипохлорита, озона, HNO3 и др.: 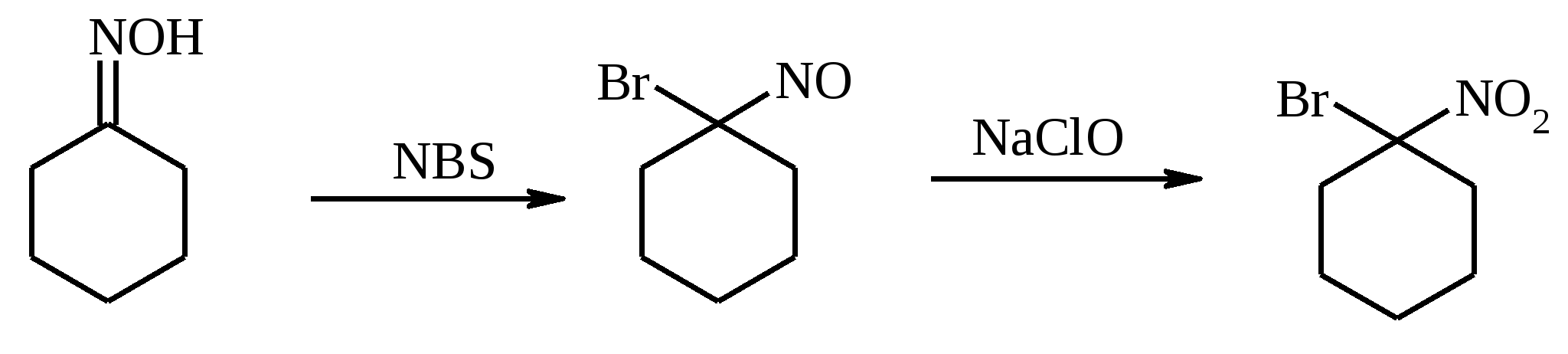 Продукты аналогичного строения могут быть получены в одну стадию при галогенировании кетоксимов бромом или хлором в щелочной среде: 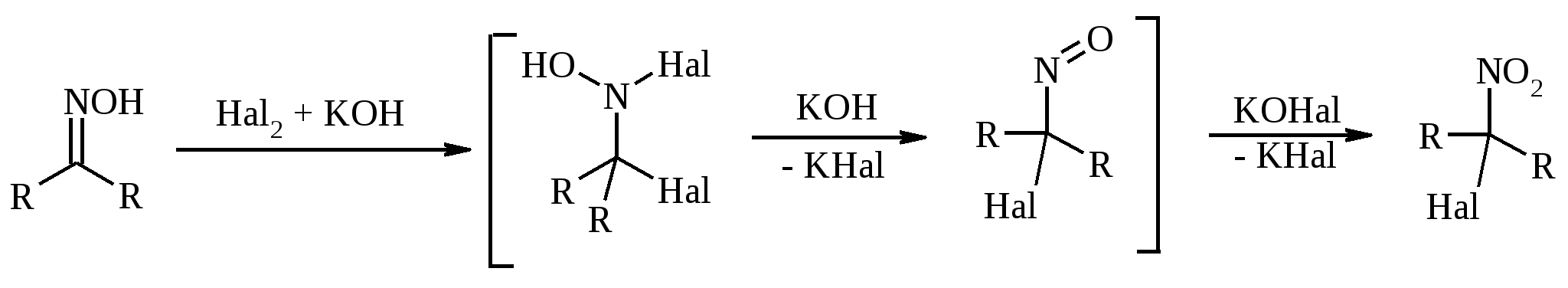 Азосоединения окисляются в азоксипроизводные перкислотами или гидропероксидами в присутствии комплексов молибдена:  16. СИНТЕЗ НЕКОТОРЫХ ОКИСЛИТЕЛЕЙ 16.1. ДИХРОМАТ ПИРИДИНИЯ 2CrO3 + 2C5H5N + H2O → (C5H5NH)2Cr2O7 К 200 г (2.00 моль) оксида хрома (VI) в 200 мл воды прикапывают при перемешивании и охлаждении льдом 158 г (2.00 моль, 161 мл) пиридина. После этого прибавляют 800 мл ацетона и смесь охлаждают до -20ºС. Через 3 ч выпавшие оранжевые кристаллы отфильтровывают, промывают их ацетоном и сушат при комнатной температуре под вакуумом, получая 312 г (82%) продукта с т. пл. 149ºС. Дихромат пиридиния – удобный в обращении нейтральный окислитель, использование которого позволяет превращать спирты в альдегиды и карбоновые кислоты. 16.2. ХЛОРХРОМАТ ПИРИДИНИЯ 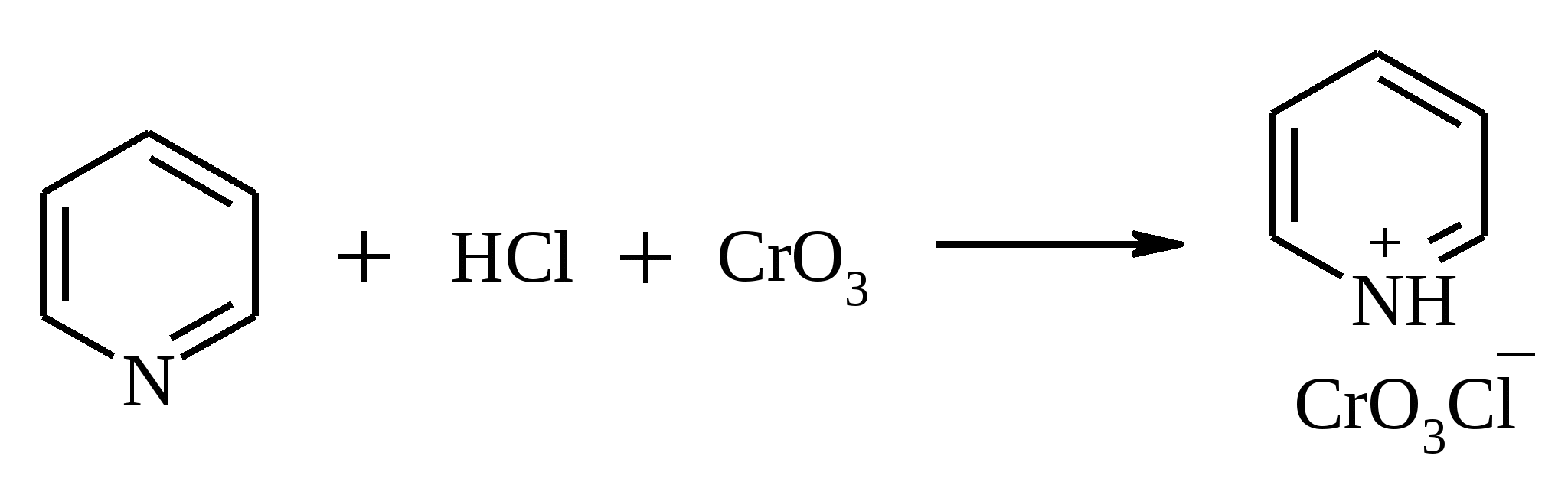 25 г (0.25 моль) оксида хрома (VI) при интенсивном перемешивании вносят в 46 мл 6 М соляной кислоты. Через 3 мин раствор охлаждают до 0ºС и в течение 10 мин при перемешивании прикапывают 19.7 г (0.25 моль, 20 мл) пиридина. Хлорхромат пиридиния, выпавший в виде оранжевых игл, быстро отфильтровывают на стеклянном фильтре и сушат 1 ч под вакуумом над оксидом фосфора, получая 45.2 г (84%) продукта. Реагент необходимо хранить в темноте без доступа влаги. Хлорхромат пиридиния – хороший реагент для окисления первичных и вторичных спиртов до альдегидов и кетонов соответственно, обладает слабокислыми свойствами. 16.3. ХЛОРИСТЫЙ ХРОМИЛ CrO3 + 2 HCl = CrO2Cl2 + H2O К раствору 50 г (0.5 моль) CrO3 и 170 мл конц. HCl при охлаждении льдом прибавляют порциями по 20 мл 100 мл конц. H2SO4. Смесь жидкостей переливают в делительную воронку; через 20 мин сливают нижний слой CrO2Cl2 в круглодонную колбу со шлифом. Через жидкость в течение нескольких минут пропускают сухой воздух и затем перегоняют неочищенный CrO2Cl2 при атмосферном давлении в хорошо высушенном приборе на шлифах. Хлористый хромил (диоксид-дихлорид хрома(VI)) – темно-красная жидкость, сильно дымящая во влажном воздухе. Хранят ее в запаянных стеклянных сосудах в темноте; т. пл. -96.5ºС, т. кип. 117ºС, d425 1.91; растворим в CCl4, CHCl3, C6H6, POCl3. |