Глик Молекулярная биотехнология. Глик Б., Пастернак Дж. Молекулярная биотехнология. Принципы и применение. Пер с англ. М. Мир, 2002. 589 с
 Скачать 9.74 Mb. Скачать 9.74 Mb.
|

ГЛАВА 8 |
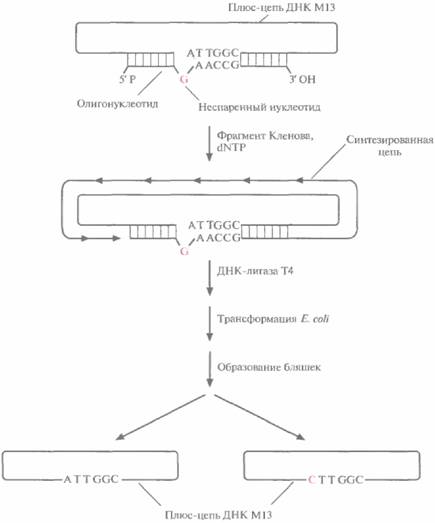 | Рис. 8.1. Олигонуклеотид-направленный мутагенез. Одноцепочечную ДНК фага М13 (плюс-цепь), несущую ген-мишень, отжигают с комплементарным синтетическим олигонуклеотидом, содержащим одно основание, не комплементарное соответствующему основанию исходной ДНК, Олигонуклеотид служит затравкой для синтеза ДНК, а М13-вектор с встроенным геном — матрицей. Репликацию катализирует фрагмент Кленова ДНК-полимеразы E E. coli. Синтезированную полноразмерную цепь замыкает в кольцо ДНК-лигаза Т4. Образовавшимися двух цепочечными молекулами трансформируют E. coli. Часть фаговых частиц содержит ДНК дикого типа, часть — мутантную ДНК. |
частицы, что в конечном счете приводит к лизису клеток и образованию бляшек. Поскольку репликация идет по полуконсервативному механизму, половина популяции образующихся фаговых частиц должна содержать ДНК дикого типа, а половина — мутантную ДНК со специфической нуклеотидной заменой. Частицы, содержащие только мутантный ген, идентифицируют при помощи ДНК-гибридизации в жестких условиях, используя в качестве зонда исходный олигонуклеотид. Мутантный ген вырезают и встраивают в какой-либо экспрессирующий Е. соli-вектор. Мутантный белок синтезируют в E. coli и очищают.
На самом деле число фаговых частиц, несущих мутантную ДНК, оказывается гораздо меньше ожидаемых 50%: лишь 1—5% бляшек содержат фаг с мутантным геном. Чтобы повысить выход мутантного фага, метод олигонуклеотиднаправленного мутагенеза модифицировали. Один из подходов состоял во введении М13-вектора, несущего ген, в который необходимо внести мутацию, в штамм E. coli, дефектный по двум ферментам метаболизма ДНК (рис. 8,2). Один фермент — это мутантная форма dUTP-пирофосфатазы (dut). Клетки с неактивной dUTP-пирофосфатазой характеризуются повышенным содержанием dUTP, что приводит к
Направленный мутагенез и генная инженерия белков 161
встраиванию в ДНК при репликации нескольких остатков dUTP вместо dTTP. Второй фермент – это дефектная урацил-К-гликозилаза (ung). В отсутствие функциональной урацил-М-гликозилазы остатки dUТР, случайно встроившиеся в ДНК, не могут быть удалены. В одноцепочечной ДНК M13, синтезированной в таких клетках Е. соli, примерно 1% тимидиновых остатков оказываются замененными уридиновыми. Олигонуклеотид с некомплементарным основанием отжигают с урацилсодержащей ДНК М13 и in vitro достраивают вторую
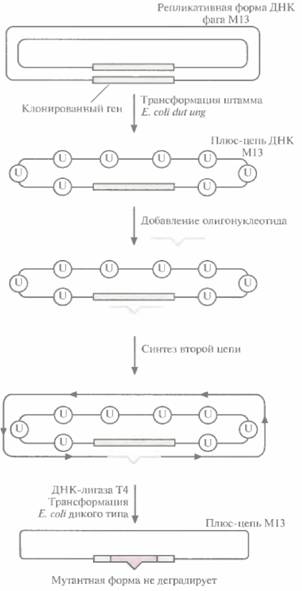 | Рис. 8.2. Повышение выхода мутантного фага M13 путем трансформации штамма Е. coltdia ung. Ген-мишень встраивают в двух цепочечную репликативную форму ДНК фага MI3 и полученными молекулами трансформируют штамм Е. coli dut ung. Мутация dut вызывает повышение содержания dUTP в клетке, что приводит к включению в ДНК нескольких остатков dUTP (U). а мутация ung блокирует их удаление. Двухцепочечной ДНК М13, содержащей ген-мишень, трансформируют клетки E. coli дикого типа. Продукт гена ung дикого типа (урацил-N-гликозилаза) удаляет все остатки урацила из исходной цепи, и она деградирует. Мутантная цепь остается интактной, поскольку она не содержит остатков урацила. Эта цепь служит матрицей для репликации ДНК, и в результате доля фаговых частиц, несущих мутантный ген, увеличивается. |
цепь. Двухцепочечной ДНК трансформируют штамм E. coli, содержащий функциональный ген ung. Активная урацил-N-гликозилаза хозяйских клеток удаляет остатки уридина из ДНК M13 (рис. 8.2), исходная матричная цепь М13 деградирует и далее реплицируется только мутантная цепь, не содержащая dUTP, В результате выход фаговых частиц, несущих мутантный ген, значительно увеличивается.
Олигонуклеотид-направлепный мутагенез с использованием плазмидной ДНК
Основной недостаток одигонуклеотид-направленного мутагенеза с использованием фага М13 — большое число процедур. Чтобы выделить мутантную форму нужного гена, приходится затратить много времени. В качестве альтернативы системе с использованием фага M13 было разработано множество других подходов, основанных на применении плазмидных ДНК. Это позволяет обойтись без переноса интересующего исследователя гена из плазмиды в фаговую ДНК, а после завершения мутагенеза — обратно в плазмиду. Один из этих подходов включает встраивание ДНК в плазмидный вектор, который несет функциональный ген устойчивости к тетрациклину и неактивный ген устойчивости к ампициллину; в середине последнего заменен один нуклеотид (рис. 8.3). Клетки E. coli трансформируют вектором, несущим ДНК-мишень, и двухцепочечную плазмидную ДНК денатурируют щелочью с тем, чтобы получить одноцепочечные кольцевые молекулы. Денатурированную ДНК отжигают с тремя разными олигонуклеоти-
162 ГЛАВА 8]
дами. Один из них предназначен для внесения изменений в клонированную ДНК-мишень, второй — для устранения мутации в гене устойчивости к ампициллину, третий — для замены одного нуклеотида в гене устойчивости к тетрациклину с тем, чтобы инактивировать этот ген. В реакционную смесь добавляют четыре дезоксирибонуклеозидтрифосфата и ДНК-полимеразу Т4, функционирующую аналогично фрагменту Кле-нова ДHК-полимеразы I Е. coli. Гибридизовавшиеся олигонуклеотиды служат затравками для синтеза ДНК, а интактная кольцевая молекула
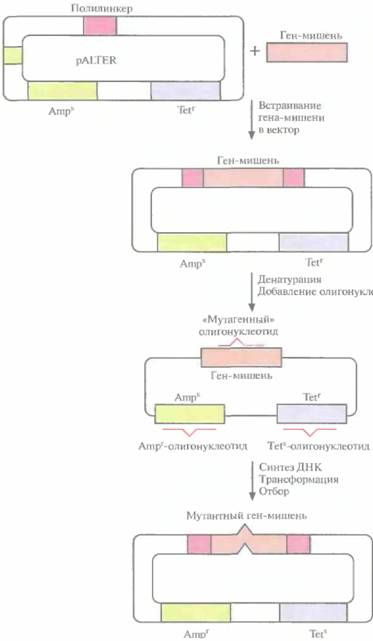 | Рис, 8.3. Олигонуклеотид-направленный мутагенез с использованием плазмидной ДНК. Ген-мишень встраивают в полилинкер вектора pALTER. Плазмидную ДНК денатурируют в щелочи и отжигают с тремя олигонуклеотидами: «мутагенным» олигонуклеотидом, олигонуклеотидом, восстанавливающим устойчивость к ампициллину (Ampr), и олигонуклеотидом, придающим чувствительность к тетрациклину (Tets). Эти олигонуклеотиды служат затравками для синтеза ДНК с помощью ДНК-полимеразы Т4, а исходная цепь — матрицей. Одноцепочечные разрывы в новосинтезированной цепи зашиваются ДНК-лигазой Т4. Продуктами реакции трансформируют клетки E. coli и отбирают трансформантов Аmрг и TetS. |
Направленный мутагенез и генная инженерия белков 163
ДНК — матрицей. Одноцепочечные разрывы в новосинтезированной цепи зашиваются с помощью ДНК-лигазы Т4. По окончании синтеза и лигирования продуктами реакции трансформируют клетки E. coli. Трансформантов отбирают по признаку устойчивости к ампициллину и чувствительности к тетрациклину. Примерно 90% из них содержат специфическую мутацию в клонированном гене. У остальных трансформантов клонированный ген не был изменен либо потому, что олигонуклеотид не гибридизовался с ним, либо потому, что он вытеснялся в ходе синтеза ДНК. Клетки, несущие мутантный клонированный ген, идентифицируют с помощью гибридизации. Все плазмиды, штаммы, ферменты, олигонуклеотиды (кроме того, который предназначен для изменения клонированного гена), а также буферы продаются в наборе, что облегчает работу.
Олигонуклеотид-направленный мутагенез с использованием ПЦР-амплификации
Более простой и быстрый метод получения больших количеств мутантных генов, альтернативный системе с использованием фага М13, -сайт-специфический мутагенез в сочетании с полимеразной цепной реакцией (ПЦР), Один из вариантов этого подхода состоит в следующем. Ген-мишень встраивают в плазмидный вектор (рис. 8.4) и помешают препарат в две пробирки. В каждую из них добавляют по два специфических праймера для ПЦР: 1 и 2 в одну пробирку, 3 и 4 — в другую. Праймеры 2 и 3 полностью комплементарны одному из участков клонированного гена или прилегающей к нему последовательности, а 1 и 3 комплементарны другому участку, но содержат один некомплементарный нуклеотид и гибридизуются с разными цепями, так что в результате происходит замена обоих нуклеотидов данной пары. Положение сайтов гибридизации праймеров l и 2 в одной пробирке и 3 и 4 — в другой таково, что ΠЦР-продукты в разных пробирках имеют разные концы. По окончании ПЦР содержимое пробирок объединяют и проводят денатурацию, а затем ренатура-цию. Поскольку концы амплифицированных молекул ДНК из двух пробирок неодинаковы, одноцепочечные ДНК из разных пробирок ассоциируют с образованием кольцевых молекул с двумя одноцепочечными разрывами. Эти разрывы репарируются in vivo после трансформации E. coli. При ренатурации одиночных цепей из одной пробирки образуются линейные молекулы. В клетках E. coti стабильно поддерживаются в виде плазмид и наследуются только кольцевые, а не линейные молекулы, при этом все они несут сайт-специфическую мутацию. Таким образом, с помощью описанного метода можно вносить точковые мутации в клонированный ген, при этом отпадает необходимость во встраивании гена в ДНК фага M13, использовании мутантных штаммов Е. coli типа dut ung и в переносе мутантного гена из М13-вектора в экспрессирующий вектор.
Случайный мутагенез с использованием «вырожденных» олигонукмотидных праймеров
К сожалению, обычно бывает неизвестно, какую нуклеотидную замену в клонированном гене нужно произвести, чтобы получить белок с нужными свойствами. Поэтому часто приходится изменять один определенный нуклеотидный сайт всеми возможными способами. Например, можно синтезировать олигонуклеотидные праймеры, в одном из сайтов которых находятся разные нуклеотиды. Такие «вырожденные» олигонуклеотиды обычно получают, добавляя в автоматический синтезатор ДНК на определенном этапе, когда к цепи должен просоединяться специфический нуклеотид, небольшое количество (до нескольких процентов) трех других нуклеотидов (рис. 8.5). В результате получается гетерогенный по одному сайту набор олигонуклеотидных праймеров, с помощью которых можно получить соответствующий набор мутантных генов-мишеней с нуклеотидными заменами в специфическом сайте.
Этот подход имеет два преимущества: 1) не нужно в точности знать, какую роль играет тот или иной аминокислотный остаток в функционировании белка: 2) поскольку в данном сайте происходят разные аминокислотные замены, могут случайно синтезироваться белки с разнообразными интересными и полезными свойствами. Конечно, если ни один из образующихся белков не обладает нужными свойствами, приходится все начинать сначала, синтезировав новый набор "вырожденных" праймеров, комплементарных другой области гена.
164 ГЛАВА 8
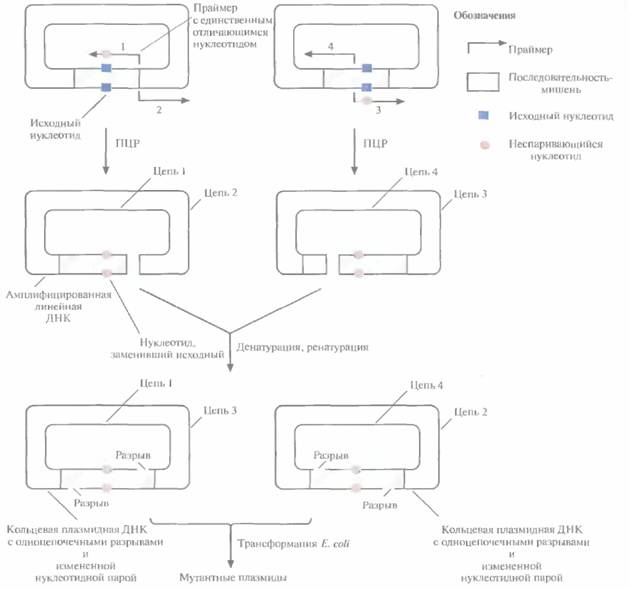 |
| Рис. 8.4. Олигонуклеотид-направленный мутагенез с использованием ПЦР. Реакцию проводят в двух пробирках, в каждой из которых содержится одинаковая двухце π очечная плазмидная ДНК, но разные наборы праймеров. Πраймеры 1 и 3 содержат один неспаривающийся нуклеотид и комплементарны разным цепям плазмидной ДНК. Праймеры 2 и 4 полностью комплементарны соответствующим участкам плазмидной ДНК и тоже гибридизуются с разными цепями. Положение сайтов гибридизации для праймеров каждой пары различается, но их концы стыкуются. В результате ПЦР-амплификации образуются линейные молекулы. По окончании реакции содержимое пробирок смешивают и проводят денатурацию, а затем ренатурацию. В результате кроме двух исходных линейных амплифицированных молекул образуются две кольцевые плазмидные ДНК, каждая с двумя одноцепочечными разрывами. После трансформации кольцевыми молекулами клеток Е. coliразрывы репарируются ферментами клетки-хозяина, и плазмида может реплицироваться независимо. Линейные молекулы ДН К в E. coli не сохраняются. |