Глик Молекулярная биотехнология. Глик Б., Пастернак Дж. Молекулярная биотехнология. Принципы и применение. Пер с англ. М. Мир, 2002. 589 с
 Скачать 9.74 Mb. Скачать 9.74 Mb.
|

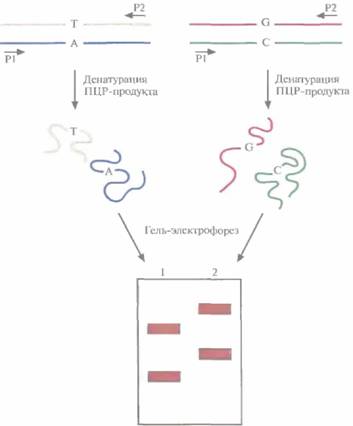
Клонирование генов заболеваний человекаКак правило, ген конкретного заболевания человека нельзя клонировать, руководствуясь каким-то заранее составленным набором экспериментальных протоколов. Выбор имеющихся в распоряжении исследователя методов и средств зависит от конкретных условий. Начало поиска гена заболевания определяется имеющейся информацией о продукте данного гена. В одних случаях генный продукт бывает хорошо известен, в других можно лишь догадываться, что он собой представляет. Наконец, для многих наследственных заболеваний природа генного продукта вообще неизвестна. Для каждого из этих случаев разработана своя стратегия. В целом для поиска гена заболевания существует четыре подхода: функциональное, кандидатное, позиционное и позиционно-кандидатное картирование. Независимо от применяемого подхода утверждать, что данный ген ассоциирован с интересующим исследователя заболеванием, можно лишь после того, как у больных обнаружены нуклеотидные изменения в гене, не встречающиеся в том же гене у здоровых индивидов. Выявление мутаций в генах человекаДля выявления мутаций разработан целый ряд простых и недорогих подходов, таких как анализ конформационного полиморфизма одноцепо-чечной ДНК (SSCP, single-strand conformational polymorphism), градиентный гель-электрофорез в денатурируют их условиях (DGGE, denaturing gradient gel electrophoresis), гетеродуплексный анализ (НА, heteroduplex analysis), химическое расщепление некомплементарных сайтов (CMC, chemical mismatch cleavage), тест на укороченный белок (РТТ, protein truncation test). Наиболее широко среди перечисленных подходов применяется SSCP. Суть метода состоит в следующем. Как можно большее (по возможности все) число экзонов исследуемого гена по отдельности амплифицируют методом ПЦР, используя в качестве матрицы ДНК больных и здоровых индивидов. Каждая пара праймеров выбирается из последовательностей, фланкирующих экзон, или из его концевых участков. Кроме того, используя данные секвенирования. выбирают праймеры для амплификации 5'-области, предшествующей первому экзону гена, 3'-области, следующей за последним экзоном, и участков, содержащих сайты сплайсинга. ПЦР-продукты каждой реакции денатурируют, быстро охлаждают и разделяют с помощью электрофореза. Благодаря внутри цепочечному спариванию комплементарных оснований и образованию других связей денатурированная од-ноцепочечная молекула ДНК принимает определенную трехмерную конформацию, зависящую от ее нуклеотидной последовательности. Вследствие комплементарности две цепи одной молекулы ДНК имеют разную нуклеотидную после- 468 ГЛАВА 20
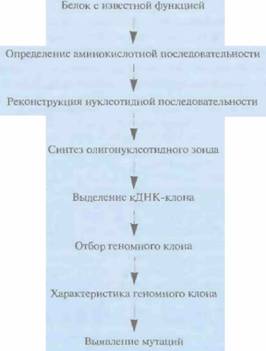
довательность, а поэтому принимают разную трехмерную конформацию и мигрируют при гель-электрофорезе с разной скоростью, В результате после разделения в геле наблюдаются две полосы, отвечающие разным комплементарным цепям. Если две молекулы ДНК, представляющие один и тот же участок гена, но полученные из разных источников, различаются одной парой нуклеотидов, то с большой вероятностью конформации одиночных цепей таких молекул ДНК будут различаться. Другими словами, каждая из четырех цепей будет перемещаться при гель-электрофорезе со своей скоростью (рис. 20,22). С помощью метода SSCP можно лишь локализовать нуклеотидные изменения в определенном экзоне или специфической области гена, но не определить природу мутации; такую информацию может дать лишь секвенирование. Метод SSCP имеет свои ограничения: он выявляет около 90% однонуклеотидных изменений в ПЦР-продуктах длиной не более 200 п. н. Функциональное картированиеФункциональное картирование гена начинается с определения аминокислотной последовательности белка с известной функцией, что позволяет реконструировать нуклеотидную последовательность кодирующей области соответствующего гена (гена-мишени). Основываясь на этих данных, синтезируют олигонуклеотидные зонды и проводят скрининг кДНК-библиотеки, полученной для ткани, в которой данный белок присутствует в большом количестве. Если можно получить очищенную мРНК, с которой транслируется данный белок, то на ней как на матрице можно синтезировать полноразмерную кДНК и клонировать ее. Правильность выбора или синтеза кДНК-клона проверяют секвенированием. Хромосомную локализацию гена-мишени определяют методом гибридизации in situ с кДНК-клоном или выбранным с его помощью геномным клоном. Для более точной локализации Молекулярная генетика человека 469 гена-мишени можно также провести скрининг панели монохромосомных клеточных гибридов, а затем и соответствующей делеционной панели при помощи кДНК-клона или отобранного геномного клона. Затем для определения клонов, гибридизующихся с данным кДНК-клоном, проводят скрининг космцдного контига, охватывающего хромосомный район, в котором локализован ген-мишень. Отобранные геномные клоны секвенируют и, используя данные о нуклеотидной последовательности кДНК, идентифицируют экзоны, интроны и 5'-, 3'-фланкирующие последовательности гена. В отсутствие космидного контига, охватывающего район нужной хромосомы, который содержит ген-мишень, выделяют клон с крупной вставкой, содержащей данный район, при помощи кДНК- или геномного зонда. Из клона с крупной вставкой получают субклоны с небольшими вставками, и проводят их скрининг при помощи кДНК-зонда. Позитивные клоны секвенируют и характеризуют ген-мишень (рис. 20.23).
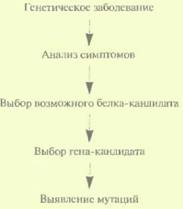
Кандидатное картированиеХотя этот подход не очень эффективен при картировании генов человека, в ряде случаев он может оказаться весьма полезным. Суть метола состоит в следующем. Анализируют симптомы генетического заболевания и на их основе пытаются понять, какого типа белок может быть с ним ассоциирован. Затем просматривают нуклеотидные последовательности всех клонированных на настоящий момент генов и выбирают ген(ы)-кандидат(ы). Основываясь на нуклеотидной последовательности гена-кандидата, вырабатывают стратегию поиска мутаций и с ее помощью пытаются установить, является ли ген-кандидат искомым геном (рис. 20.24). Принимая во внимание, что геном человека содержит очень большое число генов, а охарактеризованы лишь некоторые из них, не стоит удивляться, что правильный выбор гена случается не так уж часто. Но ценен и отрицательный результат, поскольку он позволяет исключить данный ген из числа ответственных за конкретное генетическое заболевание. Позиционное картированиеСтратегия позиционного картирования применяется в тех случаях, когда ничего не известно о продукте гена, ответственного за наследствен- 470 ГЛАВА 20 ное заболевание, и нет никаких генов-кандидатов (рис. 20.25). В подобных случаях определяют хромосомную локализацию (позицию) гена заболевания и проводят его поиск, применяя различные инструменты и средства («охота за геном»). Благоприятной для позиционного картирования является ситуация, когда у нескольких больных встречается хромосомная перестройка типа транслокации или крупной делеции (> 10 т. п. н.). Предположив, что она затрагивает ген, ответственный за патологический фенотип, при анализе сцепления используют только один специфический район хромосомы вместо того, чтобы проводить сканирование всего генома при помощи большого числа полиморфных маркеров. После локализации гена в конкретном районе хромосомы определяют его положение более точно и идентифицируют ближайшие фланкирующие маркеры, используя мультилокусное картирование с дополнительными полиморфными зондами. Минимальное расстояние между картированными маркерными сайтами, при котором их можно разграничить, в лучшем случае составляет 1 сМ, что соответствует примерно 106 п. н. На таком участке может уместиться в среднем от 20 до 50 генов. Задача позиционного картирования состоит в том, чтобы определить, какой именно из них ответствен за данное заболевание.
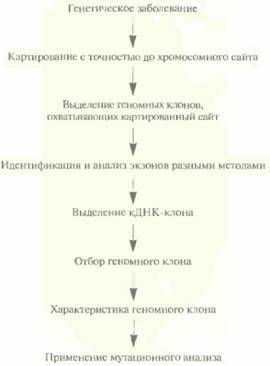
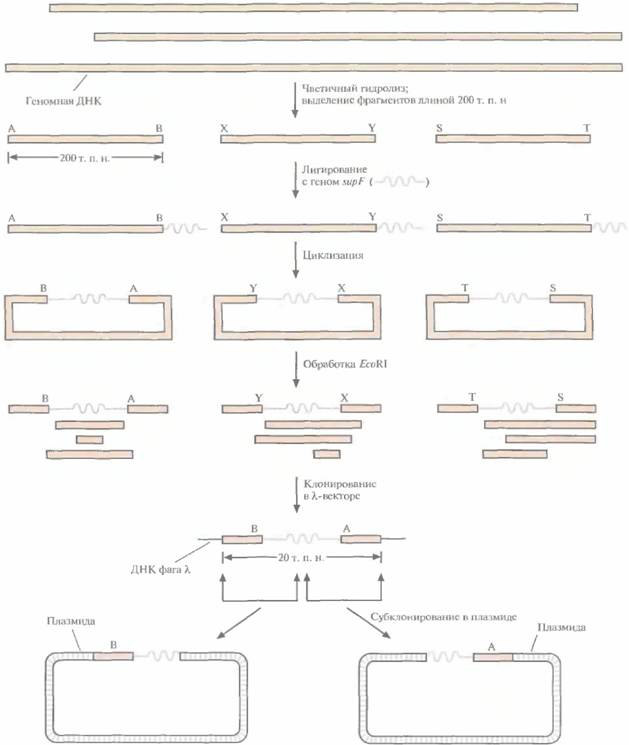
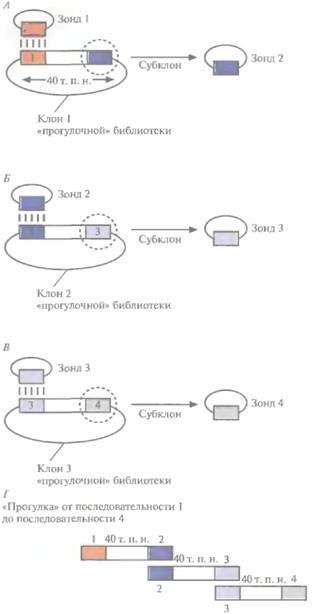
Из контига, охватывающего район хромосомы, содержащий ген заболевания, выбирают геномные клоны, которые включают фланкирующие маркеры и заключенный между ними участок ДНК. Если такой контиг отсутствует, то с помощью зондов, специфичных в отношении тесно сцепленных маркерных сайтов, проводят скрининг библиотек геномных ДНК для выявления клонов, происходящих из того района, который содержит искомый ген. Между 1986 и 1990 гг., когда метод «охоты за генами» человека еще только разрабатывался, для идентификации нужных геномных клонов использовали метод «прыжков по хромосоме» (рис. 20.26) или метод «прогулки по хромосоме» (рис. 20.27). После создания геномных библиотек, содержащих крупные фрагменты ДНК человека, и контигов эти стратегии утратили свою актуальность. Независимо от того, как именно получены нужные геномные клоны, важно знать, какие из
Молекулярная генетика человека 471
472 ГЛАВА 20
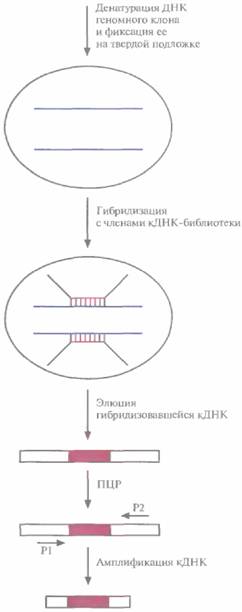
них или из субклонов содержат экзоны. Для этого можно использовать целый рад прямых и косвенных методов, таких как идентификация CpG-островков, межвидовой Саузерн-блоттинг, отбор гибридов, улавливание экзонов, секвениро-вание ДНК, компьютерный поиск. Транскрибируемым участкам геномов позвоночных часто предшествуют кластеры нуклеотидов, богатые остатками С и G (CpG-островки). Группу CpG-островков можно идентифицировать по скоплению на рестрикционной карте сайтов для рестрицирующих эндонуклеаз EagI, BssII и SacII. Если как минимум два таких сайта отделены 5—10 т. п. н. друг от друга, значит, они находятся в пределах CpG-островка. Это не гарантирует, что именно здесь находится экзон, но указывает на наличие где-то поблизости транскрибируемого гена. Молекулярная генетика человека 473 Геномные клоны или субклоны можно гибридиэовать по Саузерну с рестрицированной геномной ДНК различных позвоночных, например с ДНК мыши, крысы, кролика, обезьяны, коровы, цыпленка, рыбы (зооблот, межвидовой блоттинг, блоттинг «Ноев ковчег»). Положительная перекрестная гибридизация означает, что данный клон с большой вероятностью содержит кодирующие последовательности, поскольку многие экзоны в ходе эволюции не изменялись, в то время как повторяющиеся и некодирующие последовательности ДНК, в том числе и интроны, претерпели существенные изменения. Положительный зооблот означает, что клон содержит экзон(ы), однако не показывает, есть ли в нем ген искомого заболевания. Отбор гибридов позволяет быстро и с высокой эффективностью идентифицировать геномный клон, содержащий экзон(ы), и одновременно изолировать соответствующую кДНК, Отбор можно проводить разными способами. Обычно ДНК геномного клона из той области хромосомы, которая содержит ген заболевания, фиксируют на твердой подложке, проводят пре-гибридизацию с повторяющимися последовательностями ДНК, а затем гибридизуют с линейными векторными молекулами со вставками из кДНК-библиотеки, происходящей из ткани, вероятнее всего экспрессирующей ген-мишень. Нсгибридизовавшисся векторные молекулы смывают с фильтра, а гибридизовавшиеся элюируют и амплифицируют методом ПЦР, используя праймеры из векторных последовательностей, фланкирующих кДНК-вставку (рис. 20.28). Если точно неизвестно, в какой ткани экспрессируется ген-мишень, то кДНК-библиотеки разных тканей объединяют и проводят гибридизацию с отдельными геномными клонами. ПЦР-продукт можно затем клонировать и тестировать, с тем чтобы проверить, содержит ли он кодирующую часть гена данного заболевания. Для этого можно секвенировать кДНК и провести компьютерное сравнение нуклеотидных последовательностей этой ДНК и известных генов. Если будет получена высокая степень гомологии, можно сделать определенные выводы о том, какого типа белок кодирует данная кДНК, и если этот белок таков, что его с высокой вероятностью можно считать продуктом гена-мишени, то данный(е) клон(ы) секвенируют и идентифицируют экзоны, интроны и 5'-, 3'-фланкирующие области. Альтернативный подход состоит в поиске мутаций с целью выявления нуклеотидных различий между ДНК
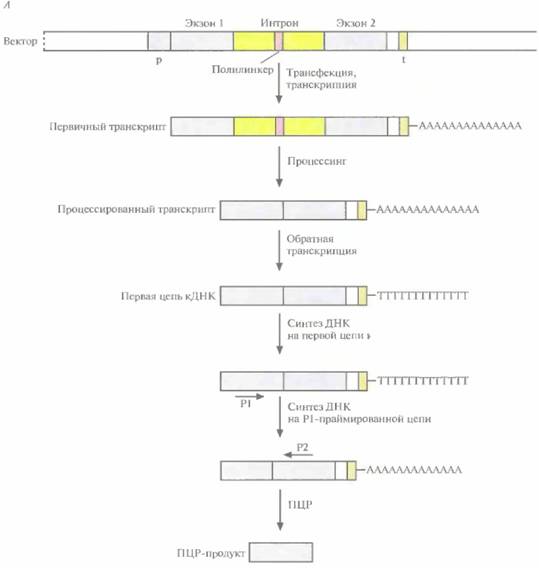
474 ГЛАВА 20 больных и здоровых индивидов. Если подход, основанный на определении степени гомологии нуклеотидных последовательностей, оказывается безуспешным, то секвенируют и анализируют другие гены из данной области. Реально при поиске гена-мишени для экономии времени и средств характеризуют в первом приближении сразу несколько генов, пока не найдут наиболее вероятный ген-кандидат, который исследуют детально, в том числе с помощью мутационного анализа. Улавливание экзонов («поимка» экзонов, экзонная амплификация) — это метод, позволяющий идентифицировать и клонировать экзоны, находящиеся в субклонах, полученных из геномных клонов (рис. 20.29), Его суть состоит в
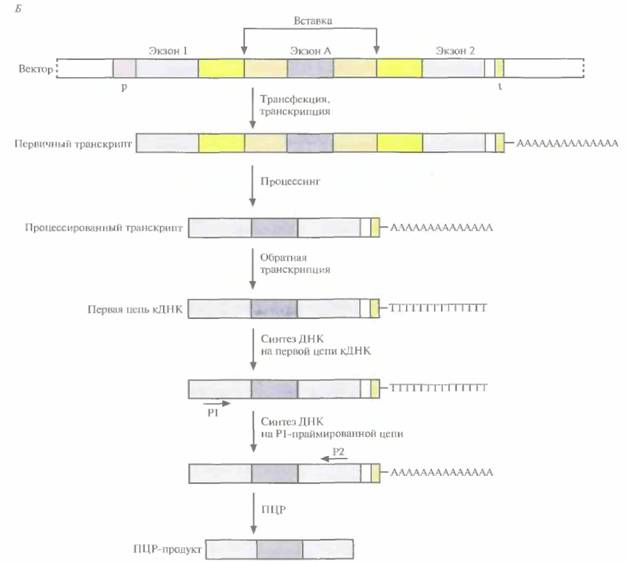
Молекулярная генетика человека 475 следующем. ДНК геномного клона расщепляют так, чтобы получить фрагменты длиной 1-6 т. п. н., и клонируют эти фрагменты в специально сконструированном векторе. Сайт множественного клонирования (полилинкер) вектора расположен внутри интрона, фланкированного двумя экзонами (экзон I и экзон 2). Этот искусственный ген (экзон 1—интрон—экзон 2) находится под контролем сильного эукариотического промотора и может реплицироваться в Е. coliили в культуре клеток млекопитающих. После введения (трансфекции) вектора без вставки в клетку млекопитающего происходит транскрипция искусственного гена и удаление интрона из пер-
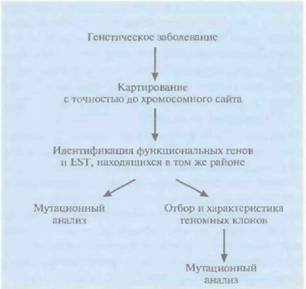
476 ГЛАВА 20 винного транскрипта. Процессированную РНК (экзон 1 —экзон 2) можно выявить, проведя ΠЦР-амплификацию обратного транскрипта. Сначала с помощью обратной транскриптазы на мРНК синтезируют ДНК (синтез первой цепи). Вторую цепь синтезируют при участии праймера, комплементарного части экзона 1 первой цепи. Затем в реакционную смесь добавляют второй праймер, комплементарный части экзона 2 второй цепи ДНК, и проводят ПЦР-амплификацию. Длину ПЦР-продукта определяют с помощью гель-электрофореза. Если в вектор встроить рестрикционный фрагмент, содержащий некий экзон А и фланкирующие его интроны, то после трансфекции процессированный транскрипт будет содержать три экзона: экзон 1—экзон А—экзон 2, Длина ПЦР-продукта будет больше, чем в тех случаях, когда в векторе нет вставки, когда вставка содержит экзон без функциональных сайтов сплайсинга (донорного и акцепторного) или когда вставка вообще не содержит экзона. Если во вставке присутствует более одного экзона, каждый из которых имеет функциональные сайты сплайсинга, то процессированный транскрипт будет содержать все эти экзоны. В том случае, если в каждом праймере содержатся рестрикционные сайты, клонируют ПЦР-продукт, несущий «пойманный» экзон, и используют последний в качестве зонда для скрининга кДНК-библиотеки. Зная нуклеотидную последовательность «пойманного» экзона, предпринимают поиск гомологичных ему последовательностей в базе данных. Если есть основания полагать, что «пойманный» экзон с большой вероятностью является частью гена данного заболевания, то характеризуют и секвенируют геномные клоны, охватывающие место расположения данного гена, и исследуют образцы ДНК больных и здоровых индивидов с целью выявления мутаций. Поскольку мутации, ответственные за патологию, не всегда бывают равномерно распределены по всем экзонам, чем больше размер сканированной кодирующей области предполагаемого гена, тем больше вероятность обнаружения мутации. Для идентификации экзонов используют различные компьютерные программы, например GRAIL (Gene Recognition and Analysis Internet Line). Они созданы исходя из некоторых характерных для экзона особенностей. Одна из них — ожидаемая нуклеотидная последовательность кодирующей области. Если лаборатория оснащена оборудованием для широкомасштабного секвенирования, можно секвенировать геномные клоны, охватывающие область расположения искомого гена, и провести компьютерную обработку полученных данных с целью выявления экзонов. Нуклеотидную последовательность предполагаемого экзона можно использовать для поиска гомологичных ей последовательностей в генной базе данных или синтезировать на ее основе олигонуклеотидный зонд для скрининга кДНК-библиотеки, Наконец, как и в случае других методов идентификации экзонов в геномных клонах, необходимо доказать, что предполагаемый экзон является частью гена-мишени. Реализация любого проекта по позиционному картированию гена занимает много времени. За период с 1986 по 1995 г, с помощью данного подхода удалось обнаружить более 50 генов различных заболеваний человека, что можно считать большим достижением. Иногда поиск гена занимает 1—2 года, в то же время для обнаружения гена хореи Гентингтона консорциуму из нескольких исследовательских лабораторий потребовалось 10 лет. Отметим, что с клонированием все новых и новых генов и построением транскрипционных карт с высоким разрешением позиционное картирование постепенно уступает место позиционно-кандидатному. Позиционно-кандидатное картированиеПозиционно-кандидатное картирование состоит в определении хромосомной локализации гена болезни, продукт которого неизвестен, и последующем анализе современных генетических и транскрипционных карт, с тем чтобы выявить кодирующие последовательности (гены, внутригенные EST), находящиеся в этом же районе (рис. 20.30). Весьма вероятно, что одна из этих последовательностей и окажется геном данного заболевания. Если какой-либо из генов-кандидатов охарактеризован, можно провести его мутационный анализ. Как альтернативу можно использовать «кандидатные» EST в качестве зондов, отобрать с их помощью геномный клон и секвенировать его, а затем также провести му- Молекулярная генетика человека 477
тационный анализ. По мере детализации физических и транскрипционных карт позиционно-кандидатное картирование становится все более популярным при поиске генов различных заболеваний человека. Программа «Геном человека»Работа над реализацией программы «Геном человека» (HGP, Human Genome Project) официально началась 1 октября 1990 г. в США и контролирует ее Министерство энергетики (Department of Energy) совместно с Государственными институтами здоровья (National Institutes of Health) США. Ее конечная цель состоит в определении нуклеотидной последовательности всего генома человека. Полученная обширная генетическая информация станет основой для более узких проектов исследования всех моногенных генетических заболеваний и послужит трамплином для изучения сложных наследственных патологий. В 1990 г. предполагалось, что работа в рамках HGP займет 15 лет, а ее стоимость составит 2 млрд. долл. За короткое время программа стала международной, ее проекты финансируются правительствами Великобритании, Франции. Канады, Германии и Японии. В настоящее время происходит кооперация и координация усилий многих государственных и межгосударственных агентств, частных компаний и некоммерческих исследовательских институтов. HGP - обширная программа, охватывающая множество различных направлений. Та часть программы «Геном человека», которая выполняется в США, включает следующие подпрограммы: построение генетических и физических карт с высоким разрешением; снижение себестоимости и повышение эффективности крупномасштабного секвенирования ДНК; разработка новых технологий картирования генов и секвенирования ДНК; усовершенствование компьютерных технологий для обработки и хранения больших массивов данных; изучение этического, правового и социального аспектов исследований, проводимых в рамках программы. Цель последней подпрограммы состоит в создани руководств для исследователей и врачей и обосновании политики правительства, касающейся использования генетической информации. В рамках подпрограммы «Новые технологии" консорциумом нескольких групп исследователей был полностью секвенирован геном дрожжей Saccharomyces cerevisiae(150 т. п. н.). Кроме того, определены полные пуклеотидные последовательности ДНК других «модельных» организмов, таких как нематоды (Caenorhabditiselegans, 100 м. п. н.), плодовая мушка (Drosophilamelanogaster, 120 м. п. н.), бактерии (Е. coli. 4,2 т. п. н.), мышь (Mus musculus, 3000 м. п. н.). Некоторые из намеченных на период 1990—1995 гг. задач той части HGP, которая выполняется в США, в 1993 г. были пересмотрены; это связано с быстрым прогрессом в генетическом картировании благодаря внедрению микросателлитных полиморфных маркеров и построению практически полных физических карт. К 1996 г. удалось решить несколько вновь поставленных задач. Например, в 1994 г. была опубликована карта (генетического) сцепления человека, которая содержала 5826 локусов, охватывающих 4000 сМ, Хотя только для 908 локусов шансы сцепления составили больше 1000:1, ко- 478 ГЛАВА 20 нечная плотность маркеров равнялась 0,7 сМ (4000 сМ/5826 локусов), что больше ожидаемой в 1995 г. плотности карты 2-5 сМ. К 1996 г. было запланировано построение физической карты, основанной на STS, с разрешением 300 т. п. н., то есть един STS-сайт должен был приходиться на каждые 300 т. п. н. ДНК человека. Однако уже в 1995 г. была построена полная физическая карта с разрешением 200 т. п. н. Успехи в оптимизации технологии секвенирования ДНК вполне ощутимы, но не столь эффектны. Стоимость секвенирования пары оснований снизилась с 5 долл. США в 1990 г. до 0,3 долл. в 1996 г. Скорость секвенирования возросла с 10 000 оснований в день в 1990 г. до 50 000 в 1996 г. К 1998 г. предполагается секвенировать 80 м. п. н., или 2,5%, генома человека. Если не произойдет никаких кардинальных изменений, то при помощи «фабрики» из 30 автоматических секвенаторов, работающих круглые сутки, и полного набора физических карт космидных клонов можно будет секвенировать примерно 3000 м. п. н. ДНК за 6 лет, потратив на это - 900 млн. долл. США. Какие-то время и средства придется потратить еще на проверку ошибок и получение окончательной последовательности. Однако, прежде чем приступать к реализации столь крупномасштабного проекта, ученые пытаются добиться значительного повышения скорости секвенирования при помощи автоматических флуоресцентных секвенаторов, в которых используется метод Сэнгера. Кроме того, предпринимаются поиски других способов быстрого секвенирования ДНК. Чтобы регулировать работу над различными аспектами всей программы, HGP распределяет финансы между разными исследовательскими группами. В большинстве случаев ответственность за создание генетических и физических карт конкретной хромосомы делят между собой крупные центры и небольшие лаборатории, которые сотрудничают друг с другом. Некоторые из наиболее крупных исследовательских институтов занимаются укомплектованием данных о генетических и физических картах генома. В результате молекулярно-генетических исследований генома человека появляется огромное количество новых данных о полиморфных зондах, STS-клонах, содержании генетических, физических и объединенных карт, рестрикционных фрагментах, геномной дактилоскопии и нуклеотидных последовательностях ДНК. Эти данные необходимо собирать, упорядочивать, хранить, объяснять, сравнивать, объединять и предоставлять другим исследователям как в исходном, так и в окончательном виде. Эффективное использование этой информации было бы невозможно без компьютерного обеспечения, включающего в себя базы данных, системы управления базами данных, алгоритмы математического моделирования и программы автоматизации экспериментов. Область знаний, которая занимается созданием численных методов обработки информации, называется информатикой. Биоинформатика имеет дело с компьютерным анализом и управлением биологической информацией. В рамках программы HGP, касающейся усовершенствования компьютерных технологий, достигнуты значительные успехи в создании компьютерных программ, позволяющих проводить всестороннюю обработку данных по геному человека. Созданы электронные сайты, где специалисты и общественность могут получить информацию о содержании различных хромосомных карт, включая их полное графическое изображение, о методах исследования генома и программном обеспечении. Например, WWW-сайт Государственного центра по изучению генома человека в США (http://www.nhgri.nih.gov/index.html) содержит информацию о программе «Геном человека» и множество ссылок на другие центры, занимающиеся этой проблемой. С самого начала своего существования HGP должна была решать этические, правовые и социальные проблемы, связанные с картированием и секвен ированием генома человека, вырабатывать стратегию, тактику и разрабатывать законопроекты, гарантирующие ответственное использование информации по генетике человека. На самом деле HGP не ставит каких-либо принципиально новых этических, правовых или социальных вопросов, которые не возникали бы при проведении медико-генетических исследований в целом. Однако реализация HGP неизбежно приведет к идентификации большого числа генов различных заболеваний и к определению последовательности многих из них, и эта Молекулярная генетика человека 479 информация будет использоваться при разработке ДНК-диагностических тестов. Здесь возникает множество поводов для беспокойства. Не будет ли генетическая информация использоваться для дискриминации людей при медицинском страховании, приеме на работу или иммиграции? Не приведет ли ее доступность к социальному неравенству? Все ли меры приняты, чтобы сохранить конфиденциальность персональной генетической информации? Как найти баланс между нуждами личности и общества? Обладают ли частнопрактикующие врачи и врачи, работающие в клиниках, достаточными знаниями по медицинской генетике, чтобы они могли разъяснить пациентам смысл конкретного генетического теста? Сможет ли генетическое консультирование уменьшить обеспокоенность обратившегося? Не скажется ли отрицательно доступность генетической информации на семейных отношениях? Можно ли надеяться на то, что удастся получить согласие на проведение диагностического теста у достаточно осведомленного пациента? Следует ли предлагать тестирование в том случае, когда данное наследственное заболевание неизлечимо? Как повысить образовательный уровень населения, чтобы оно понимало значение генетической информации? На эти и многие другие вопросы, возникающие при изучении генетики человека, нет однозначных ответов. В США в рамках подпрограммы по изучению этического, правового и социального аспектов генетических исследований организован целый ряд мероприятий: разработаны обучающие программы, проводятся семинары и выставки для студентов, учителей, врачей, общественности, адвокатов и судей; исследована возможность генетического тестирования муковисцидоза и наследственных форм рака молочной железы, яичников и толстой кишки, созданы две комиссии (по генетической информации и страхованию; по генетическому тестированию) для исчерпывающего изучения конкретных вопросов; разрабатываются предложения для выработки федеральных законов США, которые обеспечивали бы конфиденциальность генетической информации, получаемой при идентификации личности. На основе этой программы и других исследований были сформулированы пять основных принципов, которыми следует руководствоваться при использовании генетической информации и в работе генетических консультаций: право на автономию, конфиденциальность, справедливость, беспристрастность и качество. Концепция права на автономию в данном случае означает необходимость соблюдения прав человека, обращающегося в генетическую консультацию. Например, генетическое тестирование должно проводиться добровольно и только после того, как пациент в достаточной степени информирован; тестированию должны подвергаться лишь лица, относящиеся к группе риска; тестируемые должны сами решать, будут ли они знакомиться с результатами теста. Консультируемые должны быть хорошо осведомлены о всех особенностях теста: его прогностической ценности, медицинских аспектах, характере терапии, если она возможна. Обычно считается, что генетическая информация отличается от других видов личностной информации, поэтому необходимо предусмотреть особые меры предосторожности, гарантирующие ее конфиденциальность. Справедливость и беспристрастность -- это довольно близкие понятия. Достигнуто соглашение, что генетическое консультирование должно быть доступно всем, кто в нем нуждается. Как и в случае социальных и медицинских программ, необходимо защитить права умственно неполноценных пациентов и детей. Что касается качества, то тестирование должны проводить высококвалифицированные сотрудники, используя при этом надлежащие методы и средства; все этапы должны соответствующим образом контролироваться, чтобы гарантировать правильность их использования. ЗАКЛЮЧЕНИЕИзучая родословные семей, представленных несколькими поколениями, члены которых имеют четко выраженную патологию, можно определить тип наследования многих генетических заболеваний. Зная характер наследования в семьях, можно установить, является ли данное генетическое заболевание аутосомно-доминантным, аутосомно-рецессивным, Х-сцеп- 480 ГЛАВА 20 ленным доминантным или Х-сцепленным рецессивным. В случае Х-сцепленного заболевания его ген расположен на Х-хромосоме, для ау-тосомных болезней хромосомная локализация гена неизвестна. Чтобы картировать ген в специфическом районе хромосомы, можно идентифицировать сцепленные с ним маркерные сайты, используя для этого метод ПДРФ и STRP-картирование. Для оценки сцепления между маркерным сайтом и геном заболевания используют метод максимального правдоподобия. Порядок расположения ПДРФ- и STRP-сайтов на хромосоме определяют при помощи анализа наследования гаплотипов в группе семей, представленных тремя поколениями и имеющих большое количество детей (СЕРН-семей), Кроме того, порядок расположения на хромосоме уникальных сайтов, идентифицируемых при помощи ПЦР (STS), можно проверить картированием с использованием радиационных гибридов. Физические карты хромосом (контиги) строят на основе геномных библиотек, содержащих крупные (YAC, ВАС и РАС) и небольшие (космиды, Р1 и λ) фрагменты ДНК человека, используя STS-картирование или другие подходы, в том числе геномную дактилоскопию. Транскрипционные карты состоят из участков кДНК и маркерных экспрессируемых последовательностей (EST), расположенных вдоль хромосомы. Построение генетических, физических и транскрипционных карт облегчает идентификацию и характеристику генов заболеваний. Аутентичность обнаруженного гена человека можно считать доказанной, если у больных индивидов в нем найдены изменения, отсутствующие в генах здоровых лиц. Для выявления мутаций часто используют анализ конформат шинного полиморфизма одноцепочечной ДНК (SSCP). Для идентификации нужного гена человека используют четыре метода, В первом из них, функциональном картировании, на основе данных о генном продукте синтезируют зонды для скрининга кДНК-библиотеки. Положительный кДНК-клон, содержащий кодирующую область гена-мишени, используют для отбора геномных клонов и характеристики гена в целом. Второй подход, кандидатное картирование, основывается на выборе генов, которые по имеющимся данным могут отвечать за данное генетическое заболевание. В этом случае проводят поиск мутаций в генах-кандидатах у больных и здоровых индивидов и по результатам поиска делают вывод, какой из них является геном заболевания. Третий подход, позиционное картирование, применяют в тех случаях, когда ничего не известно ни о возможном гене заболевания, ни о его продукте. Этот подход весьма трудоемок и имеет множество модификаций. Сначала, используя ПДРФ- или STRP-зонды и данные о семьях с исследуемым наследственным заболеванием, определяют район хромосомы, в котором локализован искомый ген. Затем с помощью зондов, специфичных в отношении тесно сцепленных с ним маркеров, выявляют клоны, охватывающие район локализации гена заболевания. Проверяют геномные клоны или полученные из них субклоны на наличие в них экзонов. Используя данные о нуклеотидных последовательностях различных экзонов, в той или иной степени соответствующих нуклеотидной последовательности гена заболевания, разрабатывают стратегию поиска мутаций. Четвертый подход, позиционно-кандидатное картирование, состоит в картировании гена заболевания в определенном районе хромосомы, просмотре функциональных генов и маркерных экспрессируемых последовательностей, локализованных в том же хромосомном районе, и выборе тех из них, которые могут являться искомым геном. Чтобы определить, какой именно из генов-кандидатов является таковым на самом деле, используют мутационный анализ. «Геном человека» — это широкомасштабная исследовательская программа, конечной целью которой является полное секвенирование генома человека. Различные ее направления включают построение генетических и физических карт всех хромосом человека с высоким разрешением; секвенирование геномов различных модельных организмов типа Е. coli, С. elegans, S. cerevisiae, M. musculusи A. thaliana; создание компьютерных технологий для обработки и анализа данных по генетическому и физическому картированию и секвенированию ДНК; информирование общественности по всем проблемам, связанным с получением и использованием данных по генетике человека, изучение этиче- Молекулярная генетика человека 481 ских, правовых и социальных аспектов генетических исследований. На этом пути уже достигнуты впечатляющие успехи, и есть основания полагать, что геном человека будет секвенирован к 2005 г. ЛИТЕРАТУРАBellane-Chantelot С., В. Lacroix, F. Ougen, A, Billault, S. Beaufils, S. Bertrand, L Georges, F. Gilbert, I. Gros, G. Lucotte, L. Susini, J. Codani, P. Gesnouin, S. Pook, G. Vaysseix, J. Lu-Kuo, T. Ried, D. Ward, L Chumakov, D. Le Pastier, E. Barillot, D. Cohen. 1992. Mapping the whole human genome by fingerprinting yeast artificial chromosomes. Cell 70:1059-1068. Botstein D., R. L. White, M. Skolnick, R. W. Davis. 1980. Construction of a genetic map in man using restriction fragment length polymorphisms. Am, J. Hum. Genet. 32: 314-331. Chumakov L M., P. Rigault, L Le Gall, С. Bellane-Chantelot, A. Billault, S. Guillou, P. Soularue, G. Guasconi, E. Poullier, I. Gros, M. Belova, J, Sambucy, P. Gervy, I1. Gilbert, S. Beaufils, H. Bui, C. Massart, M. De Tand, F. Dukasz, S. Lecoulant, P. Ougen, V. Perrot, M. Saunier, C. Soravito, R. Bebouaylla, A. Cohen-Akenine, E. Barillot, S, Bertrand, J. Codani, D. Caterina, I. Georges, B. Lacroix, G. Lucotte, M. Sahbatou, C. Schmit, M. Sangouard, E. Tubacher, C. Dib, S. Faure, C. Fizames, G. Gyapay, P. Millasseau, S, Nguyen, D. Muselet, A. Vignal, J. Morissette, J. Mennjnger, J. Lieman, T. Desay, A. Banks, P. Bray-Ward, D. Ward, T. Hudson, S. Gerety, S. Foote, L. Stein, D, C. Page, E. S. Lander, J. Weissenbach, D. Le Paslier, D. Cohen, 1995. A YAC contig map of the human genome. Nature (London) 377(Suppl.): 175-297. Collins F. S. 1995. Positional cloning moves from perditional to traditional. Nat. Genet. 9: 347-350. Collins F., D. Galas. 1993. A new five-year plan for the U.S. Human Genome Project. Science 262: 43-46. Cooperative Human Linkage Center (CH LC): J. C. Murray, K. H. Buetow, J. L. Weber, S. Luihvigsen, T. Sclierpbkr-llertdcnia, P. Manion, J. Quillen, V. C. Sheffield, S. Süden, G. M. Duyk; Genethon: J. Weissenbach, G, Gyapay, C. Dib, J, Morissette, G. M. Lathrop, A. Vignal; University nf Utah: R. White, N. IVfatsunami, S. Gerken, R. Mells, H. Albertsen, R. Plaetke, S. Odelberg; Yale University: D. Ward; Centre d'Etude du Polymorphisme Humain (СЕРН): J. Dausset, D. Cohen, H. Cann, 1994. A comprehensive human linkage map with centimorgan density. Science 265: 2049-2054. Сох D. R., M. Burmeister, E. R. Price, S. Kim, R. M. Myers. 1990. Radiation hybrid mapping: a somatic cell genetic method for constructing high-resolution maps of mammalian chromosomes. Science 250; 245-250. Fickett J, W. 1996, Finding genes by computer: the state of the art. Trends Genet. 12: 316-320. Green E. D., P. Green. 1991. Sequence-tagged site (STS) content mapping of human chromosomes: theoretical considerations and early experiences. PCR Methods Appi. 1: 77-90. Grompe M. 1993. The rapid detection of unknown mutations in nucleic acids. Nat. Genet. 5: 111-120. Guyer M. S., F. S. Collins. 1995. How is the Human Genome Project doing, and what have we learned so far? Proc. Natl. Acad Sei. USA 92: 10841-10848. Cyapay G., J. Morissette, A, Vignal, C. Dib, C. Fizames, P. Millasseau, S. Marc, G. Bernardi, M. Lathrop, J. Weissenbach, 1994. The 1993—1994 Genethon human genetic linkage map. Nat. Genet. 7: 246-287. Hudson T. J., L. D. Stein, S. S. Gerety, J. Ma, A. B. Castle, J. Silva, D. K. Slonim, R. Baptista, L. Kruglyak, S. Xu, X. Hu, A. M. E. Colbert, C. Rosenberg, M. P. Peeve-Daly, S. Rozen, L Hui, X. Wu, C. Vestergaan), K. M. Wilson, J. S. Bae, S, Maitra, S. Ganiatsas, C. A. Evans, M. M. DeAngelis, K. A. Ingalls, R. W. Nahf, L. T. Horton, M. O. Anderson, A. J. Collymore, W. Ye, V. Kouyoumjian, I. S. Zemsteva, J. Tarn, R. Devine, D. F. Courtney, M. T, Renaud, H. Nguyen, T. O'Connor, C. Fizames, S. Faure, G. Gyapay, C. Dib, J. Morissette, J. B. Orlin, B. W. Girren, N. Goodman, J. Weissenbach, T. L. Hawkins, S. Foote, D. C. Page, E. S. Lander. 1995. An STS-based map of the human genome. Science 270: 1945-1954. 482 ГЛАВА 20 Knoppers B. M., R. Chadwick. 1994. The Human Genome Project: under an international ethical microscope. Science 265: 2035—2036. Leppert M., V. E. Anderson, T. Quattlebaum, D. Stauffer, P. O1 Cornell, Y. Nakamura, J.-M. Lalouel, R. White. 1989. Benign familial neonatal convulsions linked to genetic markers on chromosome 20. Nature (London) 337:647-648. Ott J. 1992. Analysis of Human Genetic Linkage. Johns Hopkins University Press, Baltimore, Md. Poustka A., T. M. Pohl, D. P. Barlow, A.-M. Frischauf, H. Lehrach. 1987. Construction and use of human chromosome jumping libraries from Notl-digested DNA. Nature (London) 325: 353-355. Schüler G. D., M. S. Boguski, E. A. Stewart, L· D. Stein, G. Gyapaj, К. Rice, R. F,. While, P. Rodriguez-Tomé, A. Aggarwal, E. Bajorek, S. Bentolila, В. И. Birren, A. Butler, A. B. Castle, N. Chiannikulchai, A. Chu, C. Clee, S. Cowles, P. J. R. Day, T. Dibling, N. Drouot, I. Dunham, S. Duprat, С East, С. Edwards, J.-B. Fan, N. Fang, С. Fïzames, С. Garrett, L. Green, D. Hadley, M. Harris, P. Harrison, S. Brady, A. Hfcks, E, HoUoway, L· Hui, S. Hussain, C. Louis-DH-Sully, J. Ma, A. MacGilvery, C. Mader, A. Maratukulam, T. C. Matise, K. B. McKuskk, J. Morissette, A. Mungafl, D. Musekt, H. С Nasbaum, D. C. Page, A. Peck, S. Perkins, M. Piercy, F. Qin, .1. Quackenbush, S. Ranby, T. Reif, S. Ktizcn, C. Sanders, X. She, J. Suva, D. K. Slonim, C. Sodertund, W.-L. Sun, P. Tabar, T. Thangarajah, N. Vega-Czarny, D. Voürath, S. Voytkky, T. Wümer, X. Wu, M. D. Adams, C. Auffray, N. A. R. Walter, R. Brandon, A. Dehajia, P. N. GoodfeUow, R. Houlgatte, J. R. Hudson, Jr., S. E. Ide, K. R. lorio, W. Y. Lee, N. Seki, T. Nagase, K. Ishikawa, N. Nomura, С. Phillips, M. H. Pdymeropoulos, M. Sandusky, K. Schmitt, R. Beny, K. Swanson, R. Torres, ,ï. С Venter, J. M. Sîkela, J. S. Beckmann, J. Wcissenbach, R. M. Myers, D. R. Сох, M. R. James, D. Bentley, P. Dekukas, E. S. Lander, T. J. Hudson. 1996. Agene map of the human genome. Science 274:540—546. Taylor K., N. Hornigold, D. Conway, D. Williams, Z. Ulinowski, M. Agoehiya, P. Fattorini, P. De Jong, P. F. R. Little, J. Wolfe. 1996. Mapping the human Y chromosome by fingerprinting cosmid clones. Genome Res. 6: 235—248. Terwilliger J. D., J. Ott. 1994. Handbook of Human Genetic Linkage. Johns Hopkins University Press, Baltimore, Md. КОНТРОЛЬНЫЕ ВОПРОСЫ1. Что такое независимое наследование аллелей? 2. Нарисуйте родословную, включающую четыре поколения, у членов которой встречается заболевание, имеющее аутосомно-доминантный тип наследования. Пенетрантность заболевания составляет 70%. Что такое пенетрантностъ? Как неполная пенетрантность влияет на изучение генетических заболеваний человека? 3. Что такое лод-балл? Как он определяется? 4. Что такое генетический полиморфизм? Почему полиморфные локусы важны для картирования генов заболеваний человека? 5. Как с помощью STRP-зондов можно локализовать ген заболевания человека? 6. Что такое банк СЕРН-семей? Как он «работает»? 7. Что такое STS? Как определяют хромосомную локализацию STS? 8. Что такое контиг? Опишите подробно, как строится контиг хромосомы. 9. Как установить, что вставка геномного клона содержит кодирующую область гена? 10. Каково теоретическое обоснование позиционно-кандидатного картирования гена? |