хроматография-лекции. Хроматографические методы. Общая характеристика методов
 Скачать 6.82 Mb. Скачать 6.82 Mb.
|

|
Способы борьбы с пульсациями. 1. Применение демпфирующих устройств. Это спиральные трубки специального профиля из нержавеющей стали, включенные последовательно или параллельно в систему между насосом и дозатором. Рис. 5. Спиральный демпфер. Демпфер раскручивается при увеличении давления в нем (ускорение хода насоса). При спаде давления он скручивается, его объем уменьшается, он выдавливает из себя часть растворителя, поддерживая постоянным расход и уменьшая пульсации. Такой демпфер хо рошо работает при давлении 50 атм и выше. При давления 5-30 атм лучше сглаживает пуль сации воздушный демпфер, изготовленный из колонки (рис. 6.). Воздух в заглушенной ко лонке (6х200 мм) сжимается и пульсации гасятся. Воздух в нем растворяется за 24 часа. Рис. 6. Воздушный демпфер. 2. Применение электронных устройств. При использовании электронного датчика давления можно использовать показания датчика для управления работой насоса. При спаде давления увеличивается скорость враще ния двигателя и компенсирует уменьшение давления. Также можно скомпенсировать утечки в клапанах и частично в манжетах. Применение электронного демпфера (БПЖ-80, ХПЖ-1 и т.д.) позволяет снизить пульсации давления до 1 атм при давлении 100-150 кгс/см2. 1.6.3. Ионообменная, ионная, ион-парная хроматография. В основе методов ионообменной, ионной и ион-парной хроматографии лежит динамический процесс замещения ионов, связанных с неподвижной фазой, ионами элюента, поступающими в колонку. Основная цель хроматографического процесса разделение неорганических или органических ионов одного и того же знака. Удерживание в этих видах хроматографии определяется изменением свободной энергии реакции ионного обмена. Соотношение концентраций обменивающихся ионов в растворе и в фазе сорбента характеризуются ионообменным равновесием. Ионный обмен заключается в том, что некоторые вещества (ионообменники) при погружении в раствор электролита поглощают из него катионы или анионы, выделяя в раствор эквивалентное количество других ионов с зарядом того же знака. Между катионообменником и раствором происходит обмен катионами, между анионообменником и раствором – обмен анионами. Катионообменники представляют собой чаще всего специально синтезированные нерастворимые полимерные вещества, содержащие в своей структуре ионогенные группы кислотного характера: –SO3H; –COOH; –OH; –PO3H2; –AsO3H2. Химические формулы катионообменников схематически можно изобразить как R-SO3H; R-SO3Na. В первом случае катионообменник находится в Н-форме, во втором в Na-форме. R – полимерная матрица. Катионообменные реакции записывают как обычные гетерогенные химические реакции: RН + Na+ RNa + H+ Анионообменники содержат в своей структуре ионогенные группы основного характера: –N(CH3)3+; =NH2+; =NH+ и др. Их химические формулы могут быть изображены как RNH3OH и RNH3Cl или ROH, RCl. В первом случае анионообменник находится в ОН-форме, во втором – в Сl-форме. Анионообменную реакцию можно записать следующим образом: R – OH + Cl– RCl + OH– Известны амфотерные ионообменники, содержащие в своей структуре и кислотные, и основные группы. Ионообменники, имеющие в своем составе однотипные (например, SO3H) кислотные (основные) группы, называют монофункциональными; ионообменники, содержащие разнотипные (например, SO3H, ОН) кислотные (основные) группы полифункциональными. Монофункциональные ионообменники получают реакцией полимеризации. Реакция поликонденсации позволяет получать полифункциональные ионообменники. Для того, чтобы полученные ионообменники имели достаточно высокие эксплуатационные характеристики, они должны быть нерастворимыми, но набухающими в соответствующем растворителе и иметь достаточно большое количество ионогенных групп, способных к обмену с ионогенными группами анализируемой пробы. Это может быть достигнуто, если полученные полимерные цепи достаточно разветвлены и связаны друг с другом «сшивающими мостиками». Например, при получении катионообменников полимеризационного типа на основе стирола в качестве сшивающего агента чаще всего используется дивинилбензол, введение которого в количестве до 16% обеспечивает получение ионообменников с различной степенью набухания и, следовательно, позволяет регулировать пористость ионообменника. Степенью набухания ионита, выражаемой в миллилитр/грамм, называют объем упакованного в колонку 1 г воздушно-сухого ионообменника. Содержание в ионообменнике ионогенных групп, способных к обмену с ионогенными группами анализируемой пробы, определяет так нназываемую обменную емкость ионита. Обменную емкость ионита выражают в миллиэквивалентах или миллимолях обмениваемых ионов на 1 г сухого или 1 мл набухшего ионообменника. Ионообменник поглощает, как правило, один из противоионов ионов, находящихся в подвижной фазе, т. е. проявляет определенную селективность. Экспериментально установлены ряды сродства, или селективности, ионов по отношению к ионообменникам разных типов. Например, при низких концентрациях раствора на сильнокислотных катионообменниках ионы с одинаковым зарядом сорбируются в такой последовательности: Li+ Na+ K+ Rb+ Cs+ Mg2+ Ca2+ Sr2+ Ba2+. Для ионов с разными зарядами сорбируемость увеличивается с увеличением заряда: Na+ Ca2+ < Al3+ < Th4+. Однако изменение условий проведения реакции ионного обмена может привести к обращению ряда. Ряды сродства установлены и для анионообменников. Например, сорбируемость анионов на сильноосновных анионитах увеличивается в ряду: F– OH– Cl– Br– NO3– J– SCN– ClO4–. Ионообменики, содержащие в своей структуре сильнокислотные или сильноосновные группы, вступают в реакции ионного обмена с любыми ионами, находящимися в растворе обладающими зарядами того же знака, что и знак противоиона. Такие ионообменники называют универсальными. Процесс ионного обмена между анализируемым веществом и ионообменником может быть осуществлен одним из трех способов: статическим, динамическим (способ ионообменного фильтра) и хроматографическим. Статический метод ионного обмена заключается в том, что навеску ионита приводят в контакт с определенным объемом раствора и перемешивают или встряхивают определенное время до установления равновесия. Это быстрый и простой способ ионного обмена, применяющийся для концентрирования ионов из разбавленных растворов, удаления ненужных примесей, но он не обеспечивает полного поглощения ионов, так как ионный обмен это неравновесный процесс, и вследствие этого не гарантирует полного разделения ионов. При проведении ионного обмена динамическим способом через колонку с ионитом пропускают раствор, который по мере перемещения по колонке контактирует с новыми гранулами ионита. Этот процесс обеспечивает более полный обмен, чем статический метод, так как продукты обмена удаляются потоком раствора. Им можно концентрировать ионы из разбавленных растворов и разделять ионы, сильно различающиеся по свойствам, например, разнозарядные ионы (отделять катионы от анионов), но разделение ионов одного знака заряда практически невозможно. Количественное разделение таких ионов возможно только при многократном повторении сорбционно-десорбционных элементарных актов в динамических условиях, т. е. хроматографическим методом. При работе этим методом применяют высокие слои ионита и в этот слой вводят разделяемую смесь в количестве, значительно меньшем емкости колонки, благодаря чему и обеспечивается многократное повторение элементарных актов ионного обмена. По технике проведения анализа ионообменная хроматография сходна с молекулярной и может осуществляться по элюентному (проявительному), фронтальному и вытеснительному вариантам. Отличие между молекулярной и ионообменной хроматографией состоит в том, что в молекулярной хроматографии разделенные компоненты смеси элюируются из колонки чистым элюентом, а в ионообменной в качестве элюента используют раствор электролита. При этом обмениваемый ион элюента должен сорбироваться менее селективно, чем любой из ионов разделяемой смеси. При проведении проявительной ионообменной хроматографии, которая применяется наиболее часто, колонку, заполненную ионитом, сначала промывают раствором электролита до тех пор, пока в ионите не произойдет полное замещение всех его ионов на ионы, содержащиеся в элюенте. Затем в колонку вводят небольшой объем раствора анализируемого вещества, имеющего в своем составе разделяемые ионы в количестве около 1% от емкости ионита. Далее колонку промывают раствором элюента, отбирая фракции элюата и анализируя их. Смесь ионов Cl–, Br–, J– можно разделить на высокоосновном анионите (сшитый полистирол, содержащий группы четвертичных аммониевых оснований N (CH3)3+), например, AB-17, имеющем ряд избирательности (селективности): NO3– Cl– Br– J–. Вследствие этого в качестве элюента используется раствор NaNO3. Вначале через ионит пропускается этот раствор до полного насыщения ионами NO3–. При введении в колонку разделяемой смеси ионы Cl–, Br–, J– поглощаются анионитом, вытеснив ионы NO3–. При последующем промывании колонки раствором NaNO3 ионы Cl–, Br–, J– в верхних слоях анионита постепенно вновь замещаются ионами NO3–. Быстрее всех будут вытесняться ионы Cl–, дольше всех в колонку задержатся ионы J–. Различие в селективности ионита к ионам смеси приводит к тому, что в колонке образуются отдельные зоны сорбированных ионов Cl–, Br– и J–, перемещающиеся по колонке с различной скоростью. По мере перемещения по колонке расстояние между зонами увеличивается. В каждой зоне находится лишь один из анионов разделяемой смеси и анион элюента, в промежутке между зонами лишь анион элюента. Таким образом, в элюенте на выходе из колонки будут появляться фракции, в которых содержатся отдельные компоненты разделяемой смеси. Для решения практических задач варьируют условия разделения ионов, подбирая подходящую подвижную фазу (состав, концентрация, рН, ионная сила) или изменяя пористость полимерной матрицы ионита, т. е. число межцепных связей в матрице, и создавая ионитовые сита, проницаемые для одних ионов и способные к их обмену и непроницаемые для других. Можно также изменять природу и взаимное расположение ионогенных групп, а также получать сорбенты, способные к селективным химическим реакциям за счет комплексообразования. Высокой селективностью обладают, например, комплексообразующие ионообменники, содержащие в своей структуре хелатообразующие группы органических реагентов диметилглиоксима, дитизона, 8-оксихинолина и др., а также краун-эфиры. Наибольшее применение в ионообменной, ионной и ион-парной хроматографии находят синтетические макро- и микросетчатые органические ионообменники, имеющие большую обменную емкость (3–7 ммоль/г), а также неорганические ионообменные материалы. Микросетчатые ионообменники способны к обмену ионов только в набухшем состоянии, макросетчатые – в набухшем и ненабухшем состояниях. Другим структурным типом ионообменников являются поверхностно-пленочные иониты, твердая сердцевина которых изготовлена из непористого сополимера стирола и дивинилбензола, стекла или силикагеля и окружена тонкой пленкой ионообменника. Общий диаметр такой частицы составляет около 40 мкм, толщина пленки ионита – 1 мкм. Недостаток таких ионообменников – сравнительно большой диаметр частиц и малая обменная емкость из-за низкой удельной поверхности, вследствие чего приходится работать с малыми пробами и, соответственно, использовать высокочувствительные детекторы. Кроме того, такие ионообменники достаточно быстро отравляются и не способны к регенерации. В высокоэффективной ионообменной и ионной хроматографии применяют объемно-пористые полистирольные ионообменники, объемно-пористые кремнеземы с диаметром гранул около 10 мкм, а также практически не набухающие поверхностно-пористые и поверхностно-модифицированные сополимеры стирола и дивинилбензола с ионогенными сульфо- и аминогруппами. В ион-парной хроматографии используют «щеточные» сорбенты – силикагели с привитыми обращенными фазами С2, С8 ,С18, которые легко превращаются в катионообменник при поглощении из подвижной фазы ионогенных поверхностно-активных веществ, например алкилсульфатов или солей четвертичных аммониевых оснований. При проведении хроматографического разделения с применениием ионообменников в качестве подвижной фазы чаще всего используют водные растворы солей. Это связано с тем, что вода обладает прекрасными растворяющими и ионизирующими свойствами, благодаря чему молекулы анализируемой пробы мгновенно диссоциируют на ионы, ионообменные группы ионообменника гидратируются и также переходят в полностью или частично диссоциированную форму. Это обеспечивает быстрый обмен противоионов. На элюирующую силу подвижной фазы основное влияние оказывает рН, ионная сила, природа буферного раствора, содержание органического растворителя или поверхностно-активного вещества (ион-парная хроматография). Значение рН выбирают в зависимости от природы ионогенных групп, разделяемых ионов и матрицы. С сильнокислотными и сильноосновными ионообменниками можно работать при рН = 2–12, со слабокислотными при рН = 5–12, со слабоосновными при рН = 2–6. Сорбенты на основе кремнезема при рН 9 использовать нельзя. Ионная сила подвижной фазы влияет на емкость ионообменника. С увеличением ионной силы сорбция ионов обычно уменьшается, так как растет элюирующая сила подвижной фазы. Поэтому в начале разделения подвижная фаза должна иметь малое значение ионной силы (0,05–0,1), а конечное значение этой характеристики не должно превышать 2. При градиентном элюировании часто используют буферы с увеличивающейся ионной силой. Для селективного элюирования ионов, поглощенных ионообменником, можно применять воду, буферные растворы (фосфатный, ацетатный, боратный, гидрокарбонатный и др.) с определенным значением рН и ионной силы, растворы минеральных (соляная, азотная, серная, фосфорная) и органических (фенол, лимонная, молочная, винная, щавелевая, ЭДТА) кислот. Выбор элюента облегчается тем, что предельные коэффициенты распределения большинства элементов между водными (водно-органическими) растворами многих комплексантов и ионообменниками стандартного типа определены и представлены в таблицах. 1.6.4. Эксклюзионная хроматография. Эксклюзионная хроматография это разновидность жидкостной хроматографии, в которой разделение компонентов основано на распределении молекул в соответствии с их размером между растворителем, находящимся в порах сорбента, и растворителем, протекающим между его частицами. В процессе разделения небольшие молекулы попадают в сетку полимера, в порах которой растворитель служит неподвижной фазой, и удерживаются там. Большие молекулы не могут проникнуть в полимерную сетку и вымываются из колонки подвижной фазой. Вначале элюируются самые большие, затем средние и, наконец, небольшие молекулы. 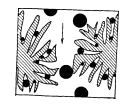 Эксклюзионная хроматография подразделяется на гель-про-никающую и гель-фильтрационную. В гель-проникающей хроматографии разделение происходит на полимерах, набухающих в органических растворителях. Гель-фильтрационный вариант эксклюзионной хроматографии предполагает использование в качестве неподвижных фаз полимеров, набухающих в воде. Продолжительность удерживания компонентов анализируемой пробы в эксклюзионной колонке зависит от размеров их молекул и диффузии в поры сорбента, а также от размеров пор неподвижной фазы. В этом виде жидкостной хроматографии коэффициент распределения D для самых маленьких молекул анализируемой пробы, которые движутся в хроматографической колонке с наименьшей скоростью, проникая в сетку неподвижной фазы, равен 1, так как подвижная фаза и растворитель, находящийся в порах неподвижной фазы, имеют один и тот же состав. При этом основное уравнение колоночной хроматографии приобретает вид 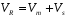 . .Молекулы большого размера, не попадающие в поры неподвижной фазы, элюируют из колонки вместе с подвижной фазой. Для них D = 0, a VR = Vm. Такой диапазон значений коэффициента распределения (от 0 до 1) характерен только для эксклюзионной хроматографии. Все молекулы анализируемого многокомпонентного вещества должны вымываться из колонки при пропускании небольшого объема растворителя от Vm до Vm + Vs и разделение заканчивается до выхода пика растворителя. Поэтому в этом виде хроматографии необходимо использовать достаточно длинные колонки с большим свободным объемом Vm и большим числом пор в сорбенте. Разрешение хроматографических пиков при эксклюзионном разделении может быть улучшено при использовании градиентного элюирования смешанными растворителями. Каждый сорбент, применяемый в эксклюзионной хроматографии, характеризуется определенным объемом пор и, следовательно, обладает определенной областью разделяемых молекулярных масс и определенным градуировочным графиком. При этом градуировочный график, характеризующий зависимость удерживаемого объема от молекулярной массы или размера молекул, имеет, как правило, сложный вид. Неподвижные фазы в эксклюзионной хроматографии выбирают исходя из конкретных аналитических задач. Первоначально устанавливают, какая система растворителей может быть использована для анализа (водная или водно-органическая). В зависимости от этого определяют тип сорбента. Если необходимо провести разделение водорастворимых проб, в качестве неподвижных фаз применяют, например, набухающие в воде сшитые декстраны (сефадексы) или полиакриламиды (биогель Р). Разделение веществ, растворимых в органических растворителях, можно проводить на полистиролах с различной степенью сшивки, набухающих в органических растворителях (стирогель, порагель, биобид С). Такие набухшие гели, как правило, неустойчивы к давлению, при их использовании допускаются очень низкие скорости потока подвижной фазы, что увеличивает время анализа. Чтобы осуществить высокоэффективный вариант эксклюзионной хроматографии, необходимо применять неподвижные фазы с жесткими матрицами силикагели, недостаток которых высокая адсорбционная активность – устраняется силанизацией поверхности или подбором соответствующего по полярности элюента. В качестве подвижных фаз в эксклюзионной хроматографии могут использоваться вещества, которые: полностью растворяют анализируемый образец; хорошо смачивают сорбент; противодействуют адсорбции компонентов пробы на сорбенте; имеют низкую вязкость и токсичность. 1.6.5. Плоскостная хроматография. К плоскостной хроматографии относятся тонкослойная и бумажная хроматографии. Эти виды жидкостной хроматографии просты по технике выполнения, экспрессны, не требуют дорогостоящего оборудования, что является их неоспоримым достоинством. Разделение смеси веществ этими методами может быть выполнено с использованием различных хроматографических систем. Поэтому выделяют адсорбционную, распределительную, нормально- и обращенно-фазовую, ионообменную и т. п. бумажную и тонкослойную хроматографии. В настоящее время наибольшее распространение получила тонкослойная хроматография. Бумажная и тонкослойная хроматографии сходны по технике выполнения. В качестве неподвижной фазы в бумажной хроматографии применяется целлюлозное волокно бумаги, в тонкослойной хроматографии различные сорбенты (Al2O3, силикагель и др.), нанесенные равномерным тонким (100300 мкм) слоем на стеклянную, металлическую или пластиковую подложку (носитель). Слой адсорбента на носителе может быть закреплен или не закреплен. Хроматографическое разделение в плоскостных методах, как и на колонке, обусловлено переносом компонентов анализируемого вещества подвижной фазой вдоль слоя неподвижной фазы с различными скоростями в соответствии с коэффициентами распределения разделяемых веществ. В обоих случаях используются хроматографические системы жидкость твердый сорбент (адсорбционный механизм разделения), жидкость жидкость твердый носитель (распределительный, ионообменный и другие механизмы). В качестве подвижных фаз применяют различные растворители или их смеси, органические или неорганические кислоты. Практическое получение плоскостных хроматограмм состоит в следующем. На полоске хроматографической бумаги или на тонком слое сорбента карандашом отмечают стартовую линию на расстоянии 1 см от нижнего края полоски или пластинки. Микропипеткой наносят пробу на линию старта в виде пятна диаметром не более 23 мм. Затем край полоски или пластинки опускают в сосуд с подвижной фазой, находящийся в герметичной камере. По мере подъема подвижной фазы по полоске или пластинке и протекания обычных в хроматографии многократных элементарных актов сорбции-десорбции, распределения между двумя жидкими фазами, ионного обмена и др. происходит разделение компонентов анализируемой смеси. Процесс обычно продолжают до тех пор, пока растворитель не пройдет от линии старта 10 см. После этого полоску или пластинку извлекают из камеры и высушивают. Если компоненты анализируемого вещества окрашены, они дают на хроматограмме соответствующие цветные пятна. Для обнаружения неокрашенных компонентов анализируемого вещества хроматограмму необходимо проявить. Проявление хроматограммы и детектирование компонентов пробы может быть проведено различными методами и зависит от состава анализируемых смесей. Проявление может быть осуществлено: с помощью УФ-освещения. Метод применим для обнаружения веществ, способных под действием УФ-излучения испускать собственное излучение (люминесцировать) видимого диапазона длин волн; посредством реагентов-проявителей. Например, присутствие в анализируемой смеси аминокислот может быть обнаружено с помощью нингидрина. Высушенную хроматограмму погружают в 0,2%-ный раствор нингидрина в ацетоне, затем высушивают ее. Пятна, соответствующие различным компонентам смеси, приобретают визуальную и, как правило, специфичную для каждого вещества окраску; с использованием иода. При этом детектируемую хроматограмму вносят в сосуд, на дне которого находятся кристаллы иода. Пары иода адсорбируются на пятнах сильнее, благодаря чему пятна визуализируются. Иод это неспецифический реагент-проявитель. Используя специфические реагенты, можно не только определить количество компонентов смеси, но и идентифицировать разделенные вещества по цвету пятен. Бумажную и тонкослойную хроматографии чаще всего осуществляют в так называемом восходящем варианте, описанном выше. Достаточно часто для улучшения качества хроматограмм приходится использовать и более сложные варианты плоскостной хроматографии, например, нисходящую, круговую, двухмерную. При проведении нисходящей бумажной или тонкослойной хроматографии анализируемое вещество наносится на стартовую линию пластинки или бумажной полоски, находящейся сверху, и элюент подается не снизу, а сверху. Положительный эффект, заключающийся в улучшении разделения, обусловлен вкладом в процесс разделения сил тяжести компонентов. L2 фронт старт L1 L Рис. 1.10. Вид и характеристики тонкослойной хроматограммы Как восходящая, так и нисходящая хроматографии могут быть осуществлены в одно и двухмерном вариантах. В отличие от описанного выше одномерного процесса разделения в плоском слое при двухмерном хроматографическом разделении разделение анализируемой пробы сначала проводят в одном растворителе, затем осуществляют разделение в направлении, перпендикулярном первому, с использованием другого растворителя, повернув первую хроматограмму на 90оС. При проведении круговой хроматографии анализируемое вещество наносится в виде капли в середину пластинки или листа хроматографической бумаги. Сюда же каплями подается один или несколько растворителей. Это приводит к тому, что получаемая хроматограмма представляет собой набор радиальных пятен. Положение пятен (зон), которые образуют разделенные компоненты анализируемого вещества на плоской хроматограмме, характеризуется величинами относительной скорости перемещения компонентов в тонком слое Rfi . Экспериментально величину Rfi определяют как отношение расстояния Li, пройденного i-м компонентом, к расстоянию L, пройденному растворителем от стартовой линии до линии фронта (рис. 1.10): 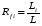 (1.41) (1.41)Величина Rfi зависит от природы соответствующего компонента анализируемой пробы, природы неподвижной фазы, ее толщины, природы и качества подвижной фазы, способа нанесения пробы и других факторов, но всегда Rfi 1. Величина Rfi фактически тождественна времени удерживания вещества или его удерживаемому объему, характеризующим скорость прохождения вещества через хроматографическую колонку, и может быть использована для качественной идентификации компонентов анализируемой пробы, а диаметр пятна тождественен высоте или площади хроматографического пика и, следовательно, в некоторой степени отражает количественное содержание вещества. Количественное определение состава анализируемой пробы в простейшем случае может быть оценено визуально по интенсивности собственной окраски пятен или интенсивности флуоресцентного свечения полученных пятен при УФ-детектировании. Для этих целей достаточно широко применяется элюирование хроматографических пятен. При этом пятно, полученное на хроматограмме, аккуратно вырезают или соскребают, обрабатывают подходящим растворителем и полученный раствор исследуют соответствующим физико-химичес-ким методом. Можно использовать и весовой метод, при котором соответствующее пятно вырезают из хроматограммы и взвешивают. Количество вещества определяют по разности весов чистой бумаги такой же площади и бумаги с веществом. Бумажная (БХ) и тонкослойная хроматография (ТСХ) по механизму разделения относятся к распределительной хроматографии. В методе БХ носителем является специальная хроматографическая бумага с определенными свойствами. Неподвижной фазой служит вода, адсорбированная на поверхности и порах бумаги (до 20%), подвижной органический растворитель, смешивающийся или несмешивающийся с водой, вода или растворы электролитов. Механизм хроматографического разделения на бумаге довольно сложен. В неподвижной фазе вещество может удерживаться не только вследствие растворения в адсорбированной бумагой воде, но и адсорбироваться непосредственно целлюлозой. Нанесенные на бумагу разделяемые компоненты переходят в подвижную фазу и по капиллярам бумаги перемещаются с различными скоростями в соответствии с коэффициентом межфазного распределения каждого из них. В начале хроматографирования некоторая часть вещества из бумаги переходит в подвижную фазу и перемещаются далее. Когда органический растворитель достигает участка бумаги, не содержащего растворенное вещество, снова происходит перераспределение: из органической фазы вещество переходит в водную, сорбированную на бумаге. Поскольку компоненты обладают различным сродством к сорбенту, при перемещении элюента происходит разделение: одни вещества задерживаются в начале пути, другие продвигаются далее. Здесь сочетаются термодинамический (установления равновесного распределения веществ между фазами) и кинетический (движение компонентов с различной скоростью) аспекты разделения. В результате каждый компонент концентрируется на определенном участке бумажного листа: образуются зоны отдельных компонентов на хроматограмме. Использование хроматографии на бумаге имеет ряд существенных недостатков: зависимость процесса разделения от состава и свойств бумаги, изменение содержания воды в порах бумаги при изменении условий хранения, очень низкая скорость хроматографирования (до нескольких суток), низкая воспроизводимость результатов. Эти недостатки серьезно влияют на распространение хроматографии на бумаге как хроматографического метода. В методе ТСХ процесс разделения смеси веществ осуществляется в тонком слое сорбента, нанесенного на инертную твердую подложку, и обеспечивается движением подвижной фазы (растворителя) через сорбент под действием капиллярных сил. По механизму разделенияразличают распределительную, адсорбционную и ионообменную хроматографию. Разделение компонентов происходит в этих случаях либо в результате их различного коэффициента распределения между двумя жидкими фазами (распределительная хроматография), либо вследствие различной адсорбируемости соединений сорбентом (адсорбционная хроматография). Адсорбционный метод основан на разной степени сорбции-десорбции разделяемых компонентов на неподвижной фазе. Адсорбция осуществляется за счет ван-дер-ваальсовских сил, являющейся основой физической адсорбции, полимолекулярной (образование нескольких слоев адсорбата на поверхности адсорбента) и хемосорбцией (химического взаимодействия адсорбента и адсорбата). В случае использования для ТСХ таких сорбентов, как окись алюминия или силикагель в разделении играют роль как распределение, так и адсорбция на развитой активной поверхности сорбента (150 750 м2/г). Распределение компонентов смеси происходит между водой на поверхности носителя (такие адсорбенты, как окись алюминия, крахмал, целлюлоза, кизельгур – и вода образуют неподвижную фазу), и перемещающимся через эту неподвижную фазу растворителем (подвижная фаза). Компонент смеси, легче растворимый в воде, перемещается медленнее, чем тот, который легче растворим в подвижной фазе. Адсорбция проявляется в том, что между носителем, например, окисью алюминия, и компонентами смеси устанавливаются адсорбционные равновесия – для каждого компонента свое, результатом чего является разная скорость перемещения компонентов смеси. Можно выделить два крайних случая: а) концентрация вещества на адсорбенте равна нулю. Вещество полностью растворяется в подвижной фазе и увлекается ею (перемещается вместе с фронтом растворителя). б) вещество адсорбируется полностью, с растворителем не взаимодействует и остается на старте. На практике при умелом подборе растворителя и адсорбента распределение соединения располагается между этими крайними случаями, и вещество постепенно переносится от одного слоя сорбента к другому за счет одновременно происходящих процессов сорбции и десорбции. Растворитель, проходящий через сорбент, называют элюентом, процесс перемещения вещества вместе с элюентом элюированием. По мере продвижения жидкости на пластинке происходит разделение смеси веществ благодаря действию сил адсорбции, распределения, ионного обмена или совокупности действия всех перечисленных факторов. В результате образуются отдельные хроматографические зоны компонентов смеси, т.е. получается хроматограмма. Правильный подбор сорбента и элюента определяет эффективность разделения смеси. Подвижность исследуемого вещества зависит от его сродства к сорбенту и элюирующей силы (полярности) элюента. С увеличение полярности соединения растет и его сродство к полярному сорбенту. По увеличению степени адсорбции силикагелем органические соединения располагаются в ряд: углеводороды <алкилгалогенидыарены<нитросоединения<простые эфиры <сложные эфиры<альдегиды<спирты<амины<карбоновые кислоты. В свою очередь для силикагеля элюенты можно расположить в порядке возрастания «полярности» (элюирующей способности) и сформировать серию растворителей (элюотропный ряд) в соответствии с экспериментальными данными: алканы>бензол>хлороформ>диэтиловый эфир> этилацетат>спирты С2-С4>вода>ацетон>уксусная кислота>метанол. Таким образом, полярное соединение – спирт достаточно сильно адсорбируется на силикагеле и поэтому слабо перемещается под действием такого неполярного растворителя, как гексан, и остается около линии старта. В свою очередь неполярный ароматический углеводород бифенил заметно более подвижен в гексане, но даже здесь для достижения Rf около 0,5 необходим более полярный апротонный элюент – хлористый метилен. Силу элюента регулируют, используя смеси растворителей – соседей по элюотропному ряду с разной полярностью. В настоящее время в ТСХ применяют главным образом следующие сорбенты: для разделения липофильных веществ силикагель, окись алюминия, ацетилированную целлюлозу, полиамиды; для разделения гидрофильных веществ целлюлозу, целлюлозные ионообменники, кизельгур, полиамиды. Важнейшей характеристикой сорбента является его активность, т.е. способность сорбировать (удерживать) компоненты разделяемой смеси. За рубежом ряд фирм производит силикагель, кизельгур и окись алюминия с добавкой 5% гипса, который используется для закрепления слоя сорбента при самостоятельном изготовлении пластин. Наиболее распространенным сорбентом является силикагель - гидратированная кремниевая кислота, образующаяся при действии минеральных кислот на Na2SiO3 и сушкой образовавшегося золя. После размалывания золя используют фракцию определенной зернистости (указанную на пластинке, обычно 5-20 мкм). Силикагель является полярным сорбентом c группами ОН в качестве активных центров. Он легко сорбирует на поверхности воду и образует водородные связи. Окись алюминия является слабоосновным адсорбентом и используется в основном для разделения соединений слабоосновного и нейтрального характера. Недостатком пластин на окиси алюминия является обязательная активация поверхности перед использованием в сушильном шкафу при высокой температуре (100150оС) и низкая, по сравнению с силикагелем адсорбционная емкость слоя. Кизельгур адсорбент, полученный из природных минералов диатомовых земель. Сорбент обладает гидрофильными свойствами и более низкой адсорбционной емкостью слоя в сравнении с силикагелем. Целлюлоза: тонкослойные пластины с нанесенной целлюлозой очень эффективны для разделения сложных органических молекул. Адсорбент представляет собой в основном шарики целлюлозы диаметром до50 мкм, закрепленные на носителе крахмалом. Как и в бумажной хроматографии, подъем фронта растворителя происходит очень медленно. Хроматографический анализ выполняется на промышленных пластинах чешского производства «Силуфол» («Silufol») из алюминиевой фольги, иногда укрепленной картоном, и «Силупласт» из пластмассы, покрытых слоем сорбентов – силикагеля LS 5-40 с крахмалом или гипсом в качестве связующего (до 10%), или оксида алюминия с добавлением и без флуоресцентных индикаторов. Пластинки «Силуфол» имеют высокую скорость элюирования, однако при этом характеризуются низкой разделяющей способностью и невысокой чувствительностью. При хранении чувствительны к условиям (влажность, температура, агрессивные среды и т.п.). Отдельные фирмы поставляют хроматографические пластинки со слоем сорбента различной (обычно до 0,25 мм), но строго постоянной толщины (силикагель, целлюлоза, ионообменная смола), на стекле и подложках из алюминиевой фольги, пластмассы, пропитанного стекловолокна. Пластины «Sorbfil» (ТУ 26-11-17-89) выпускаются в России на полимерной основе (полиэтилентерефталат, марка П) или алюминиевой подложке (марка АФ) с нанесенным рабочим слоем микрофракционированного сорбента силикагеля марки СТХ-1А и СТХ-1ВЭ (выпускался в СССР как фракционированный силикагель КСКГ) толщиной 90-120 мкм (до 200 мкм), закрепленным специальным связующим - силиказолем. При использовании в качестве связующего золя кремневой кислоты (силиказоля), который после нагревания переходит в силикагель, полученные ТСХ-пластины состоят из двух компонентов: слоя силикагеля и подложки. Равномерность по толщины слоя сорбента на одной пластине составляет ±5 мкм. Пример обозначения: "Сорбфил-ПТСХ-АФ-В-УФ (10х10)" - пластинки для ТСХ высокоэффективные на алюминиевой подложке, с люминофором, 10х10 см. Если применять стеклянную подложку (марка С), то такие пластины являются многоразовыми и химически прочными. Их химическая устойчивость определяется химической стойкостью силикагеля. В результате ТСХ-пластины могут многократно обрабатываться агрессивными реагентами, например, горячей хромовой смесью, что снимает ограничения в использовании коррелирующих реагентов для детектирования пятен и модификации сорбента, и позволяет проводить многократную (до 30 раз и более) регенерацию пластин хромовой смесью. Стеклянные пластинки могут быть нарезаны по необходимым размерам. Механическая прочность слоя сорбента может регулироваться, обеспечивая, с одной стороны, транспортировку и многократность обработки пластин и, с другой стороны, возможность экстракции слоев адсорбента с разделившимися веществами для последующего вымывания индивидуальных соединений из сорбента и их дальнейшего исследования инструментальными методами (ИК и УФ-спектрометрии, рентгено-структурными методами, ЯМР и т.д.). Пластины различаются величиной фракций (распределения частиц) силикагеля, из которого состоит слой. На аналитических пластинах (марка А) фракция 5-17 мкм, на высокоэффективных (марка В) - 8-12 мкм. Более узкое распределение повышает эффективность пластин, т.е. пятна разделяемых веществ становятся более компактными (меньшими по размерам) и поэтому лучше разделяются при прохождении фронта элюента на более короткое расстояние. На российских пластинах аналитические и высокоэффективные слои различаются не очень сильно, в отличие от пластин фирмы Merck (Германия). Применять высокоэффективные пластины нужно, если вещества не разделяются на аналитических пластинах. Выпускаются пластины всех модификаций с люминофором (марка УФ) с возбуждением 254 нм. Срок хранения не ограничен, пластины «Sorbfil» широко испытаны в анализе производных аминокислот, пестицидов, липидов, антибиотиков. Методом ТСХ осуществляется качественная идентификация компонентов. Количественное определение для ТСХ также возможно, для этого требуется нанесение точного количества вещества и дополнительные денситометрические исследования с четким фиксированием интенсивности пятен. Наиболее распространенным является полуколичественный метод. Он основан на визуальном сравнении размера и интенсивности пятна компонента с соответствующими характеристиками серии пятен этого же вещества различной концентрации (стандартные растворы сравнения). При использовании пробы в количестве 1-5 мкг таким простым методом обеспечивается точность определения содержания компонента около 5-10%. Нередко для определения компонентов в образце необходимо провести пробоподготовку для получения смеси, содержащей анализируемые соединения. Пробоподготовка основана на извлечении препаратов из образца органическими растворителями (н-гексан, петролейный эфир, диэтиловый эфир, хлороформ), очистке экстракта и последующем хроматографировании в тонком слое окиси алюминия или силикагеля. Существует несколько вариантов ТСХ и БХ, различающихся способом подачи растворителя. В зависимости от направления движения подвижной фазы различают: а) восходящую хроматографию подвижную фазу наливают на дно разделительной камеры, бумага (пластинка) ставится вертикально; б) нисходящую хроматографию подвижная фаза подаётся сверху и перемещается вниз вдоль слоя сорбента пластины или бумаги; в) радиальную хроматографию горизонтальное продвижение фронта растворителя: подвижная фаза подводится к центру бумажного диска (пластины), куда нанесена разделяемая смесь. Наиболее распространенным является восходящее элюирование (хроматографирование). Фронт элюента при этом перемещается снизу вверх. Выбор растворителя (подвижной фазы) определяется природой сорбента и свойствами разделяемых веществ. Хроматографическое разделение методами БХ и ТСХ проводят в разделительной камере с притёртой крышкой. Количественной мерой скорости переноса вещества при использовании определенного адсорбента и растворителя является величина Rf (от англ. retentionfactor – коэффициент задержки, этот параметр является аналогией времени удерживания). Положение зоны хроматографируемого компонента устанавливают по величине коэффициента Rf , равной отношению скорости движения его зоны к скорости движения фронта растворителя. Величина Rf всегда меньше единицы и не зависит от длины хроматограммы. На величину Rf оказывают влияние различные факторы. Так, при низкой температуре вещества перемещаются медленнее; загрязнения растворителей, негомогенность адсорбента, посторонние ионы в анализируемом растворе могут изменять величину Rf до 10%. В выбранной системе анализируемые вещества должны иметь различные значения Rf и распределяться по всей длине хроматограммы. Желательно, чтобы значения Rf лежало в пределах 0,05-0,85. На практике величину Rfрассчитывают как отношение расстояния l, пройденного веществом, к расстоянию L, пройденному растворителем: Rf= l / L (6.1) Обычно для расчета выбирают центр пятна (рис. 1). Величина Rfзависит от многих факторов: типа хроматографической бумаги (ее пористости, плотности, толщины, степени гидратации) и сорбента (размера зерен, природы групп на поверхности, толщины слоя, его влажности, природы вещества, состава подвижной фазы), условий эксперимента (температуры, времени хроматографирования и т.п.). При постоянстве всех параметров хроматографирования значение Rfопределяется только индивидуальными свойствами каждого компонента. 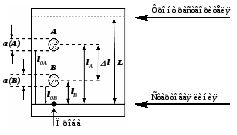 Рис. 1. Определение на хроматограмме величин Rf для компонентов А и В, степени их разделения Rs и числа теоретических тарелок N. Эффективность БХ и ТСХ также зависит от селективности и чувствительности реакций, используемых для обнаружения компонентов анализируемой смеси. Обычно используют реагенты, образующие с определяемыми компонентами окрашенные соединения проявители. Для более надёжной идентификации разделяемых компонентов применяют «свидетели» растворы стандартных веществ (в том же растворителе, что и проба), наличие которых предполагается в образце. Стандартное вещество наносят на стартовую линию рядом с анализируемой пробой и хроматографируют в одинаковых условиях. На практике часто используют относительную величину: Rf rel =Rf x / Rf stand (6.2) где Rf stand также рассчитывают по формуле (6.1). Эффективность хроматографического разделения характеризуют числом эквивалентных теоретических тарелок и их высотой. Так, в методе ТСХ число эквивалентных теоретических тарелок NА для компонента А разделяемой смеси рассчитывают по формуле: NA= 16 (lOA/ a(A))2(6.3) Значения lOAи а(А)определяют, как показано на рис. 6.1. Тогда высота эквивалентной теоретической тарелки НАсоставляет: HA= lOA / N = a(A)2 / 16lOA. (6.4) Разделение практически возможно, если Rf(А) Rf(В) 0,1. Для характеристики разделения двух компонентов А и В используют степень (критерий) разделения Rs: Rs = l / (a(A) / 2 + a(B) / 2)= 2l / (a(A)+ a(B)) (6.5) где l расстояние между центрами пятен компонентов А и В; а(А)и а(В) диаметры пятен А и В на хроматограмме (рис. 6.1). Чем больше Rs, тем чётче разделены пятна компонентов А и В на хроматограмме. Условия хроматографирования подбирают так, чтобы величина Rsотличалась от нуля и единицы, оптимальное значение Rsсоставляет 0,30,7. Для оценки селективности разделениядвух компонентов А и В используют коэффициент разделения α: |