Министерство здравоохранения республики
 Скачать 1.92 Mb. Скачать 1.92 Mb.
|

|
5. Гипо -, апластическая анемия. Эпидемиология, этиология, патогенез, клиника, диагностика, лечение Апластическая или гипопластическая анемия - гетерогенная группа заболеваний системы крови, основу которых составляет уменьшение продукции клеток костного мозга трех клеточных линий (эритроцито-, лейкоцито-, тромбоцитопоэза). Эпидемиология В литературе сведения об эпидемиологии апластических анемий малочисленны. В некоторых штатах США, например в Калифорнии, заболеваемость апластической анемией составляет примерно 1:300 000-1:500 000. Заметной зависимости заболеваемости от пола и этнической принадлежности не обнаружено. Болезнь практически может возникнуть у лиц любого возраста, однако чаще у молодых (от 20 до 40 лет). Апластическая анемия характеризуется высокой летальностью (50-75%). Этиология. Апластическая анемия может развиться при воздействии ряда миелотоксических факторов: ионизирующего излучения, химических веществ (бензол и его производные), солей золота, мышьяка, лекарственных средств-хлорамфеникола (левомицетина), фенилбутазола (бутадион), хлорпромазина (аминозин), мепробамата, дилантина, антиметаболитов (противоопухолевые средства-б-меркаптопурина, метотрексата), алкилирующих (цклофосфана, хлорбутина) и некоторых других средств. Миелотоксический эффект от воздействия одних факторов (ионизирующее излучение, антиметаболиты) возникают всегда при достаточно большой дозе, других - проявляется индивидуально. Причина индивидуальной чувствительности, в частности к некоторым лекарственным средствам не всегда ясна, но может быть связана с генетическими дефектами кроветворных клеток. Это относится, например, к хлорамфениколу и фенилбутазону, которые вызывают супрессию (в зависимости от дозы) эритропоэза с частотой соответственно1:24000 и 1:40 000 лиц, их принимающих. В 50-75% случаев причина заболевания остается неизвестной. Патогенез Патогенез до конца не изучен. Наиболее распространено мнение о поражении частично детерминированной стволовой клетки и ее микроокружения. Апластическая анемия возникает в результате глубокого угнетения кроветворения. Она может быть вызвана воздействием внешних факторов (лекарственные вещества, химические препараты, тяжелые металлы, лучистая энергия, различные инфекции), а также и внутренних (гипотиреоз, уремия). Несмотря на редкость патологии, отмечается сложность установления механизма гемолитического процесса в каждом конкретном случае, тяжесть клинико-гематологических проявлений, трудность лечения, высокой летальностью. Этиологические факторы (химические вещества сульфаниламиды, препараты бензольного ряда) оказывают повреждающее действие на хромосомный аппарат клеток костного мозга и тем самым нарушают синтез дезоксирибонуклеиновой кислоты (ДНК). Эти нарушения, в конечном счете, приводят к угнетению пролиферации костномозговых клеток. При этом костный мозг не может обеспечить необходимую выработку эритроцитов, гранулоцитов и тромбоцитов, что ведет к панцитопении. Данные изучения ранних гранулоцитарных предшественников методом культивирования клеток костного мозга в агаровом геле позволили раскрыть отдельные звенья в патогенезе синдрома гранулоцитопении. Обнаружено значительное снижение способности клеток предшественников образовывать колонии, продуцирующие клетки гранулоцитарного ряда. Обнаружено, что у части больных не существует прямой зависимости между величиной колониеобразования и количеством гранулоцитов в единице объема перифериической крови и костного мозга. Это дает основание предполагать, что одной из причин развития гранулоцитопении при апластической анемии является функциональная неполноценность стволовых клеток и неэффективный гранулоцитопоэз. Классификация апластических анемий Среди апластических анемий выделяют конституциональные и приобретенные. У детей выделяют приобретенные, семейные и врожденные формы апластических анемий. К врожденным относятся анемия Фанкони, Дайемонда-Блекфэна. Среди приобретенных анемии как результат воздействия хамических, лекарственных, инфекционных агентов и ионизирующего излучения. Часто причина остается невыясненной. Клиника Апластическая анемия может начаться остро и иметь быстро прогрессирующее течение, но чаще болезнь развивается постепенно. Клинические проявления зависят от выраженности цитопении. В клинической картине можно отметить следующие синдромы: - интоксикационный синдром; - анемический синдром; - геморрагический синдром; - септико-некротический синдром (пневмонии, абсцессы, фурункулы); - гиперпластический синдром в случае присоединения смешанного гемолиза (внутрисосудистого, внутриклеточного). При объективном исследовании отмечается бледность кожных покровов, одышка, тахикардия, выслушивается систолический шум в области сердца. Геморрагический синдром соответствует степени тромбообразования. Наблюдаются петехиальные высыпания на коже и слизистых оболочках, кровоточивость десен, носовые и маточные кровотечения. Следствием нейтропении являются инфекционные осложнения: ангины, некротические проявления на слизистых рта (Рис. 3), желудочно-кишечного тракта, пневмонии, инфекции мочевых путей, сепсис. Селезенка обычно не увеличена. При наличии гемолитического синдрома отмечается увеличение печени, селезенки и периферических лимфатических узлов. 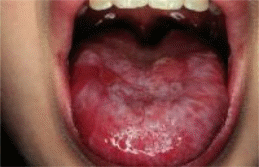 Рис. 3 - Септико-некротический синдром апластической анемии (некротический стоматит, глосит) Диагностика Картина периферической крови: выраженная анемия (концентрация гемоглобина может падать до 20-30 г/л), лейкопения (нейтропения с относительным лимфоцитозом) и тромбоцитопения, иногда до полного исчезновения тромбоцитов из крови (панцитопения - снижение всех показателей периферической крови). Анемия чаще нормохромная, макроцитарная отмечается примерно у 60-65% больных, число ретикулоцитов снижено. Содержание железа в сыворотке крови нормальное или повышенное, насыщение трансферрина близко к 99%. СОЭ обычно увеличена до 40-60 мм/час. При пункционной биопсии костного мозга (в миелограмме) отмечается малое количество ядросодержащих клеток (миелокариоцитов) или они совсем отсутствуют (трехростковая панцитопения), при гистологическом исследовании отмечаются замещение жиром деятельного костного мозга (Рис. 4).  Рис. 4 - Миелограмма при апластической анемии Дифференциальный диагноз Дифференциальную диагностику следует проводить с острым лейкозом и В12-дефицитной анемией. При этом, важное значение имеет стернальная пункция. Апластическая анемия исключается при обнаружении гиперплазии эритроцитарного ростка. Если увеличенные в количестве эритроидные элементы имеют морфологические черты мегалобластов, то вероятно наличие у больного В12-дефицитной анемии, но при этом необходимо иметь в виду и возможность эритромиелоза. Присутствие в костномозговом пунктате большого количества бластных клеток свидетельствует об остром лейкозе. Получение при пункции скудного материала еще не может быть расценено как указание на аплазию костного мозга, так как такая картина бывает и при остром лейкозе. Для уточнения диагноза производят трепанобиопсию кости, уменьшение активного кроветворного костного мозга и увеличение жировой ткани подтверждает диагноз апластической анемии. Апластическую анемию необходимо дифференцировать и с иммунной периферической цитопенией. Для диагностики иммунной цитопении имеют значение обнаружение увеличенной селезенки, положительные проба Кумбса или агрегат-гемагглютационная проба, нормальное количество мегакариоцитов в костном мозге. Дифференциальную диагностику апластической анемии проводят с пароксизмальной ночной гемоглобинурией и гемолизиновыми формами аутоиммунной гемолитической анемии. Дифференциальная диагностика основывается на отсутствии при апластической анемии признаков внутрисосудистого гемолиза, ретикулоцитоза, увеличения селезенки. Прогноз Прогноз при апластической анемии серьезный. По данным многих авторов, без проведения трансплантации костного мозга, летальность составляет 50-75%. При быстром прогрессировании болезни смерть может наступить через несколько месяцев, при хроническом течении происходит смена обострений и ремиссий. Иногда наблюдается полное выздоровление. К неблагоприятным признакам апластической анемии относятся: - постоянные трудно купирующиеся геморрагии, даже небольшие кровотечения сразу из нескольких внутренних органов, кровоизлияние в головной мозг, - глубокая нейтропения с частыми инфекционными осложнениями, возникновение сепсиса, - КОЕк ниже 25.105, - высокий стойкий относительный лимфоцитоз в гемо- и миелограмме при одновременном его выявлении, - снижение гемоглобина более чем до 50 г/л, - ретикулоцитопения, - высокая летальность (80% и более), у беременных, в период или после инфекционного гепатита. Лечение Лечение основывается на комплексной терапии и настойчивом продолжительном ее применении нередко в течение многих месяцев: - Гемотрансфузии компонентов крови (эр. масса; лейкомасса, тромбомасса). - Глюкокортикоиды (преднизолон), иногда продолжительностью до 4-6 месяцев. - Антибактериальная терапия. - Анаболические стероиды. - Витаминотерапия. - Сосудоукрепляющие средства. - Трансплантация аллогенного костного мозга. Лечение апластической анемии направлено на коррекцию цитопенического синдрома и костномозговой недостаточности и борьбу с осложнениями. Вначале отменяются все лекарственные средства, к которым у больного имеется индивидуальная повышенная чувствительность и которые могут быть причастны к развитию анемии. Консервативное специфическое лечение начинают после поступления больного в стационар и подтверждения диагноза. Для больных старше 20 лет и при легком течении заболевания консервативное лечение является основным методом лечения, поскольку аллогенная трансплантация стволовых клеток не всегда выполнима. Консервативное лечение проводят также во время подбора совместимого донора. - При тяжелой анемии проводят заместительные трансфузии крови по 150-200 мл с промежутками в 3-5 дней. Лучший эффект дают прямые переливания крови от доноров. Применяются переливания свежей эритроцитарной массы, отмытых, размороженных эритроциты. При выраженном геморрагическом синдроме показано использование концентрата тромбоцитов. - Применяют кортикостероидные гормоны: преднизолон, триамсинолон, дексаметазон, дельтакортолон, которые уменьшают тромбоцитопеническое кровотечение, удлиняют срок жизни перелитых клеток крови, снижают нежелательные реакции после трансфузии. В острой фазе необходимо назначить 60-80 мг/сут (из расчета 1 мг/кг массы) преднизолона. После наступления ремиссии доза преднизолона постепенно уменьшается. Поддерживающая доза 5-10 мг в сутки. Лечение проводится на протяжении 2-3 месяцев до исчезновения всех признаков гемолиза. - Плазмаферез целесообразно проводить с интервалом 8-10 дней на фоне продолжения приема преднизолона в дозах, что может спосоствоать уменьшению геморрагических проявлений. - Андрогены эффективны при некоторых вариантах анемии Фансони, приобретенной апластической анемии, хотя случаи успешного лечения крайне редки. - При инфекционных осложнениях применяют антибиотики широкого спектра действия. - Для повышения регенераторных возможностей костного мозга при панцитопении применяются витамины группы В. Назначаются длительными курсами по 6-8 недель В1 по 1 мл подкожно ежедневно, В2 по 5 мг 3 раза в день внутрь, В6 по 50 мг ежедневно, В12 по 100 мкг внутримышечно через день, фолиевая и пантотеновая кислота по 30 мг 3 раза в день внутрь. Аскорбиновая кислота назначается вместе с рутином. - Применяют средства, оказывающие благоприятное влияние на сосудистую стенку (дицинон, серотонин, рутин, аскорбиновая кислота), г-иминокапроновую кислоту при соответствуюших нарушениях гемостаза, андрогены (ретаболил, анаполон и др), нередко в сочетании с глюкокортикостероидами, десферал с целью уменьшения явлений гиперсидероза. - В качестве лейкостимуляторов используется пентоксил 0.2, 4-метилурацил 0.5, лейкоген 0.2, батиол 0.2 внутрь 2 раза в день (лучше со сливочным маслом), 5% раствор нуклеиновокислого натрия по 2 мл внутримышечно. - Антимоноцитарный глобулин (тимоглобулин) является препаратом выбора для лечения пациентов пожилого возраста или при отсутствии донора для трансплантации костного мозга. Назначается по 20 мг/кг/сут в день на протяжении 5 дней внутривенно капельно. Курсовая доза 160 мг/кг, - Антилимфоцитарный глобулин, по данным зарубежных авторов, использование антилимфоцитарного глобулина эффективно у 40-50% больных, что позволяет рекомендовать его на ранних этапах лечения. - Циклоспорин А (Сандиммун), почти настолько же эффективен, как тимоглобулин. Начальная доза составляет 5 мг/кг/сут внутрь или 3 мг/кг внутривенно. Далее дозы подбирают, исходя из концентрации циклоспорина в крови, определяемой ежедневно. При отсутствии эффекта в течение 120 дней препарат отменяют. - В последние годы применяют терапию колониестимулирующими фак-торами (КСФ): препараты гранулоцитарного КСФ (Г-КСФ) филграстим, ленограстим, нартограстим преимущественно стимулируют образование нейтрофилов, препараты гранулоцитарно-макрофагального КСФ (ГМ-КСФ): молграмостим, сарграмостим, лейкомакс стимулируют продукцию эозинофилов, нейтрофилов, моноцитов. Колониестимулирующие факторы назначают при неэффективности тимоглобулина или циклоспорина, применяется в сочетании с интерлейкинами. - Одним из основных методов лечения тяжелой апластической анемии является трансплантация костного мозга от доноров, подобранных по системе HLA. Трансплантация костного мозга (трансплантация стволовых клеток периферической крови или костного мозга) показана лицам молодого возраста, особенно с тяжелыми, прогностически неблагоприятными формами болезни (уровень гемоглобина ниже 20 г/л, количества нейтрофилов менее 0.5.109/л, ретикулоцитов после коррекции менее чем 1% и числа клеток костного мозга менее 25% от общего объема), а также зависимым от гемотрансфузий больным (необходимость в трансфузиях возникает чаще 1 раза в месяц). До трансплантации костного мозга проводят химиотерапию. Обычно применяется циклофосфамид в сочетании с тотальным облучением тела. 6. Анемия, обусловленная дефицитом витамина В12 Витамин В12-дефицитная анемия бала описана в 1649 году Т. Адиссоном и в 1872 году А. Бирмером. Называлась пернициозной (гибельная) до того как в 1826 г. Майно и Мерфи получили положительный результат при лечении больных сырой печенью. В 1929 г. Кастл сделал предположение, что в мясе содержится внешний фактор, а в желудочном соке - внутренний, которые образуют гемопоэтическое вещество, в 1948 г. Был открыт витамин В12-«внешний фактор» и гастромукопротеин. Эпидемиология Развитие В12-дефицитной анемии характерно преимущественно для лиц пожилого возраста (60–70 лет) и практически не встречается в детском возрасте и очень редко – у молодых женщин. По данным Herber (1985), в возрасте 30–40 лет заболевание встречается с частотой 1:500, в возрасте 60–70 лет–1:200. В странах Северной Европы (Garmell, 1996) частота распространенности В12-дефицитной анемии составляет 0,1%, среди лиц пожилого возраста–1.0%. Этиология В12-дефицитная анемия развивается в результате недостатка в организме антианемического фактора, необходимого для нормального созревания эритроцитов. Внешним фактором и является витамин В12, заболевание может развиться при нарушении всасывания витамина В12 (заболевания желудка, тонкой кишки, глистная инвазия), утилизации витамина В12 костным мозгом или при повышенном расходовании витамина В12 (беременность). Патогенез Патогенез В12-дефицитной анемии связан непосредственно с обменом витамина В12. Витамин В12 поступает в организм человека с мясом, печенью, сыром, молоком, яйцами. Находящийся в пище витамин В12 в желудке соединяется с белками «R» (Rapidbinders). Белки «R» обнаруживаются в желудочном соке, плазме, крови, слюне, грудном молоке, фагоцитах и синтезируются слюнными железами, клетками слизистых оболочек. Комплекс витамина В12 + белок «R» поступает в 12–перстную кишку, где, под влиянием протеолитических ферментов панкреатического сока, белок «R» отщепляется и освободившийся витамин В12 соединяется с гастромукопротеином, поступившим из желудка. Этот комплекс поступает в дистальный отдел тонкого кишечника, где взаимодействует со специфическими рецепторами и витамин В12 всасывается в кровь. За сутки всасывается 4–5 мкг витамина В12, что составляет 80% общего его количества, поступившего с пищей. В крови витамин В12 связывается с транспортным белком, транскобаламином–2, с помощью которого доставляется в печень и костный мозг. Витамин В12 депонируется в основном печенью (запасов хватает на 3–5 лет). Витамин В12 имеет два кофермента: дезоксиаденозилкобаламин и метилкобаламин. Дефицит метилкобаламина вызывает изменения: - нарушается превращение фолиевой кислоты в ее активную форму тетрадегидрофолиевую кислоту, которая необходима для синтеза ДНК (при дефиците витамина В12 и метилкобаламина нарушается синтез ДНК, в том числе в кроветворных клетках, главным образом в эритробластах). В результате чего эритробласты увеличиваются в объеме, ядра сохраняются, но сами клетки не созревают до зрелых эритроцитов, превращаются в мегалобласты, которые легко лизируются. - нарушается рост клеток лейкоцитарного и тромбоцитарного рядов, - нарушается образование эпителия желудочно-кишечного тракта (атрофические изменения). Дефицит дезоксиаденозилкобаламина вызывает нарушение превращения продукта метаболизма жирных кислот–метилмалоновой кислоты в янтарную. Метилмалоновая кислота токсична для нервных клеток, развивается фуникулярный миелоз (дистрофические процессы в заднебоковых столбах спинного мозга). В патогенезе заболевания имеет место: 1) нарушение секреции внутреннего фактора (атрофия слизистой желудка, гастроэктромия), 2) поражение тонкой кишки (хронический энтерит, энтеропатия, спру, резекция тонкой кишки), 3) исключение из рациона продуктов животного происхождения. 4) конкурентный расход витамина В12 (инвазия широким лентецом, синдром «слепой петли», множественный дивертикулёз тонкой кишки). 5) нарушение расщепления протеина - R в 12-перстной кишке (длительное нарушение внешнесекреторной активности поджелудочной железы). |