зайко. Н. Н. Зайко Патологическая физиология Введение Предмет и задачи патологической физиологии Патологическая физиология есть наука, изучающая жизнедеятельность больного организма. Программа
 Скачать 7.32 Mb. Скачать 7.32 Mb.
|

|
Схематически изменения в тканях при ишемии можно разделить на несколько последовательных стадий. 1. Снижение эффективности цикла Кребса, повышение интенсивности гликолиза и пентозного цикла, снижение интенсивности энергетического обмена в целом. Нарушение образования энергии на участке ишемии патогенетически связано с недостаточной доставкой кислорода и необходимых для окисления субстратов, снижением активности и синтеза ферментов, выходом ферментов из поврежденных клеток в связи с повышением проницаемости клеточных мембран, разобщением процессов окисления и фосфорилирования. 2. Истощение запасов макроэргических фосфатов сопровождается суммарным приростом содержания Ca2+ в клетке, особенно после длительных периодов ишемии. На рис. 10.3 показаны как источники и механизмы повышения цитоплазматической концентрации Са2+, так и последовательность событий, обусловленных его (Са2+) избыточным содержанием. В результате может развиться вторая фаза ишемической реакции, которая включает активацию Са2+-чувствительных протеаз и фосфолипаз, необратимые изменения структуры и целостности клеточной мембраны, потерю внутриклеточных адениновых оснований (предшественников синтеза АТФ), дальнейшее массивное поступление Са2+ из внеклеточного пространства. 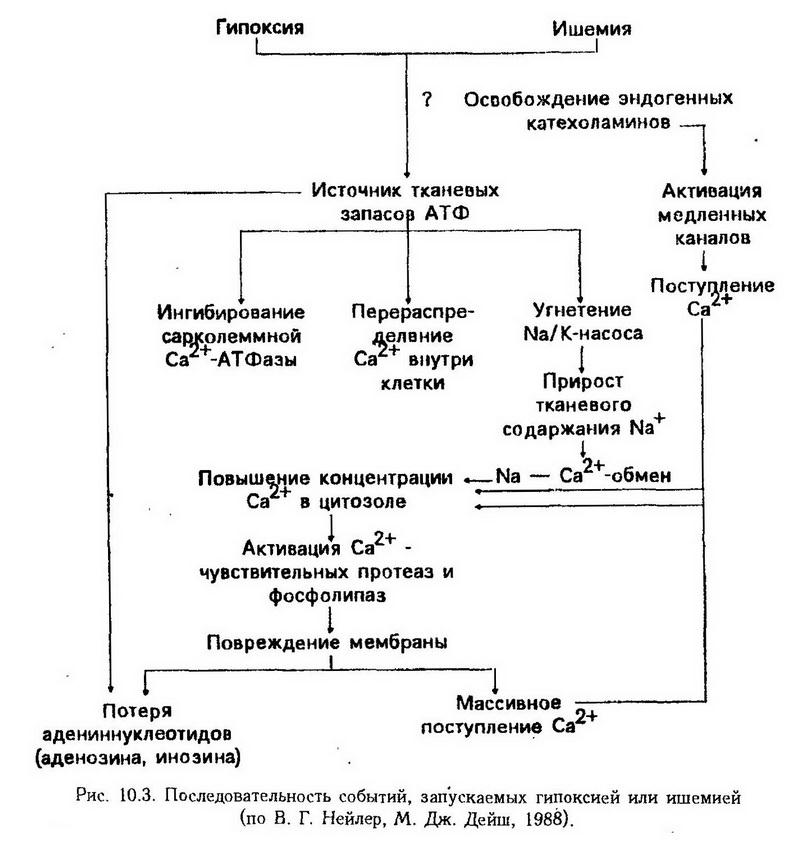 В связи с указанными изменениями нарушаются специфические функции клеток (сокращение, секреция и др.), работа ионных насосов, развивается некробиоз. Первые признаки ультраструктурных изменений обнаруживаются через несколько минут с момента возникновения ишемии и характеризуются изменениями внутренней структуры митохондрий. Наблюдается их набухание, постепенное накопление жировых капель, последующее исчезновение крист и замещение их гранулярной субстанцией. Дальнейший распад митохондрий, а также эндоплазматического ретикулума и клеточных ядер может закончиться образованием очага некроза – инфаркта. Это, как правило, происходит в паренхиматозных органах, которые отличаются повышенной чувствительностью к кислородному голоданию и особенностями ангиоархитектоники, не позволяющими достаточно быстро и эффективно устранить нарушение кровообращения вследствие развития коллатералей. Продолжительная ишемия кожи, скелетных мышц и костной ткани не вызывает столь грозных функциональных и морфологических последствий. Попытки предупредить развитие постишемических некробиотических изменений с помощью реперфузии не дали положительных результатов, поскольку в условиях реперфузии происходит дальнейшее повышение тканевого содержания кальция и усугубление повреждения мембран и митохондрий. Впервые это было установлено при ишемии миокарда. Такую реперфузию проводят под защитой антагонистов кальциевого входа. 3. Стадия усиленного биосинтеза основных биохимических компонентов соединительной ткани – коллагена, кислых и нейтральных гликозаминогликанов, являющихся основой для последующего склерозирования ишемизированного участка ткани или органа. Необходимым условием для его осуществления является усиление синтеза нуклеиновых кислот. Стаз Стаз (от грея, stasis – стояние) – замедление и остановка тока крови в капиллярах, мелких артериях и венах. Различают истинный (капиллярный) стаз, возникающий вследствие патологических изменений в капиллярах или нарушения реологических свойств крови, ишемический – вследствие полного прекращения притока крови из соответствующих артерий в капиллярную сеть и венозный. Венозный и ишемический стазы являются следствием простого замедления и остановки кровотока. Эти состояния возникают по тем же причинам, что и венозная гиперемия и ишемия. Венозный стаз может быть результатом сдавления вен, закупорки их тромбом или эмболом, а ишемический – следствием спазма, сдавления или закупорки артерий. Устранение причины стаза ведет к восстановлению нормального кровотока. Напротив, прогрессирование ишемического и венозного стазов способствует развитию истинного. При истинном стазе столб крови в капиллярах и мелких венах становится неподвижным, кровь гомогенизируется, эритроциты набухают и теряют значительную часть своего пигмента. Плазма вместе с освободившимся гемоглобином выходит за пределы сосудистой стенки. В тканях очага капиллярного стаза отмечаются признаки резкого нарушения питания, омертвение. Причиной истинного стаза могут быть физические (холод, тепло), химические (яды, концентрированный раствор натрия хлорида и других солей, скипидар, горчичное и кротоновое масло) и биологические (токсины микроорганизмов) факторы. Механизм развития истинного стаза объясняется внутрикапиллярной агрегацией эритроцитов, т. е. их склеиванием и образованием конгломератов, затрудняющих кровоток. При этом повышается периферическое сопротивление. Агрегация возникает в результате изменения физических свойств плазмолеммы эритроцитов под непосредственным действием факторов, проникающих внутрь капиллярного сосуда. При электронно-микроскопическом изучении феномена агрегации эритроцитов оказалось, что их поверхность, гладкая в нормальных условиях, становится при усиленной агрегации неровной, "пушистой". При этом изменяются сорбционные свойства эритроцитов по отношению к некоторым красителям, что свидетельствует о нарушении их физико-химического состояния. В патогенезе истинного стаза важное значение имеет замедление кровотока в капиллярных сосудах вследствие сгущения крови. Ведущую роль при этом играет повышенная проницаемость стенки капиллярных сосудов, расположенных в зоне стаза. Этому способствуют этиологические факторы, вызывающие стаз, и метаболиты, образующиеся в тканях. Особое значение в механизме стаза отводится биологически активным веществам (серотонин, брадикинин, гистамин), а также ацидотическому сдвигу тканевой реакции среды и ее коллоидному состоянию. В результате отмечается повышение проницаемости сосудистой стенки и расширение сосудов, ведущие к сгущению крови, замедлению кровотока, агрегации эритроцитов и, как следствие, – стазу. Особенно важным является выход в ткани плазменных альбуминов, способствующих понижению отрицательного заряда эритроцитов, что может сопровождаться выпадением их из взвешенного состояния. Тромбоз Тромбоз – это процесс прижизненного образования на внутренней поверхности стенки сосудов сгустков крови, состоящих из ее элементов. Сгустки крови могут быть пристеночными (частично уменьшают просвет сосудов) и закупоривающими. Первая разновидность тромбов чаще всего возникает в сердце и стволах магистральных сосудов, вторая – в мелких артериях и венах. В зависимости от того, какие компоненты преобладают в структуре тромба, различают белые, красные и смешанные тромбы. В первом случае тромб образуют тромбоциты, лейкоциты, а также небольшое количество белков плазмы; во втором – эритроциты, скрепленные нитями фибрина; смешанные тромбы представляют собой чередующиеся белые и красные слои. Еще с прошлого столетия сформировалось четкое представление об основных факторах тромбообразования в виде триады Вирхова. 1. Повреждение сосудистой стенки, возникающее под действием физических (механическая травма, электрический ток), химических (NaCl, FeCl3, HgCl2, AgNO3) и биологических (эндотоксины микроорганизмов) факторов в результате нарушения ее питания и метаболизма. Указанными нарушениями, кроме того, сопровождаются атеросклероз, гипертоническая болезнь, аллергические процессы. Пристеночный тромб образуется прежде всего на участке повреждения стенки сосуда. Это объясняется, с одной стороны, тем, что из поврежденной внутренней оболочки сосуда выделяются факторы свертывания крови, активирующие процесс тромбообразования, а с другой, локальным угнетением процесса фибринолиза, уменьшением образования в эндотелии кровеносных сосудов ПГІ2 (простациклин) и его эндоперекисей, оказывающих в норме выраженное антиагрегационное действие на тромбоциты. В условиях повреждения эндотелия увеличивается его способность к синтезу алкилсодержащих глицерофосфохолинов (фактор активации тромбоцитов – ФАТ). С его участием связывают агрегацию и дегрануляцию тромбоцитов, высвобождение из них вазоактивных аминов (гистамин, серотонин), АТФ, активацию фосфолипазы А2 и усиление биосинтеза тромбоксана А2. 2. Нарушение активности свертывающей и противосвертывающей системы крови и сосудистой стенки. Повышение активности свертывающей системы крови вследствие повышения в ней концентрации прокоагулянтов (тромбин, тромбопластин), как и понижение активности противосвертывающей (уменьшение содержания в крови антикоагулянтов или увеличение активности их ингибиторов), в том числе фибринолитической, как правило, приводит к внутрисосудистому свертыванию крови (ВССК) и тромбозу. ВССК обусловлено быстрым и значительным поступлением в сосудистое русло факторов свертывания крови, в частности тканевого тромбопластина, что наблюдается при преждевременной отслойке плаценты, эмболии околоплодными водами, травматическом шоке, остром массивном гемолизе эритроцитов. В эксперименте ВССК можно воспроизвести путем введения в систему общей циркуляции крови собак или кроликов активного тромбина или тромбопластина. ВССК может быть генерализованным (диссеминированным – ДВС-синдром) и локальным. Это процесс обратимый, особенно при своевременной терапии антикоагулянтами. Переход ВССК в тромбоз происходит под влиянием факторов свертывания сосудистой стенки и тромбоцитов при их повреждении. В связи с усиленным потреблением факторов свертывания крови и тромбоцитов в процессе ДВС, вторичной активацией противосвертывающей системы и фибринолиза во вторую фазу ДВС развивается тромбогеморрагический или гипергипокоагуляционный синдром. 3. Замедление кровотока и его нарушения (завихрения в области аневризмы). Этот фактор, вероятно, имеет меньшее значение, однако он позволяет объяснить, почему в венах тромбы образуются в пять раз чаще, чем в артериях, в венах нижних конечностей – в 3 раза чаще, чем в венах верхних конечностей, а также высокую частоту тромбообразования при декомпенсации кровообращения, пребывании на длительном постельном режиме. Процесс тромбообразования условно можно разделить на две фазы: фазу адгезии, агрегации и агглютинации тромбоцитов (клеточная фаза) и фазу коагуляции (плазматическая фаза свертывания). Физико-химическая сущность клеточной фазы (первичный или сосудисто-тромбоцитарный гемостаз) заключается в изменении электрического потенциала сосудистой стенки, заряда тромбоцитов и других клеток крови, повышении адгезивно-агрегационной способности тромбоцитов, вызывающих их оседание на поврежденной ("чужеродной") поверхности внутренней оболочки сосудов (адгезия) и "прилипание" друг к другу (агрегация). По современным представлениям, основным механизмом адгезии и агрегации тромбоцитов являются понижение их отрицательного заряда вследствие уменьшения содержания АТФ и увеличения АДФ в поврежденных участках сосудов и тканей, а также тромбоцитах, торможение антиагрегационных свойств сосудистой стенки при ее повреждении в связи с понижением синтеза ПГІ2 (простациклина) на фоне повышения синтеза в тромбоцитах ПГД2, ПГН2, тромбоксана А2, ФАТ (фактора агрегации тромбоцитов). Последние, в особенности тромбоксан А2, в противоположность простациклину являются мощными инициаторами агрегации тромбоцитов. Такое действие этих веществ, по-видимому, реализуется через высвобождение тромбоцитами АДФ, что и приводит к их агрегации. Повышенный синтез простагландинов обусловлен активирующим действием различных стимуляторов в агрегации (тромбин, коллаген, серотонин, адреналин, норадреналин и др.) на фосфолипазу А2 (высвобождающую из фосфолипидов мембран тромбоцитов арахидоновую кислоту – исходный субстрат для синтеза простагландинов), циклооксигеназу и другие ферменты, участвующие в их синтезе. Определенное участие в процессах адгезии и агрегации тромбоцитов принимают и плазменные факторы свертывания крови (фибриноген, факторы VIII, XIII). Важно отметить, что на этапе необратимой агрегации из тромбоцитов в кровь выбрасывается дополнительное количество АДФ, серотонина, адреналина, норадреналина и других биологически активных веществ, ФАТ. Процесс, таким образом, приобретает характер порочного замкнутого круга. Необратимые изменения тромбоцитов наступают через 2 – 3 мин с момента повреждения внутренней оболочки сосудов. При этом наблюдаются расширение их цитоплазмы, появление множественных псевдоподий, потеря тромбоцитарных гранул по краям агрегатов тромбоцитов, прилипание лейкоцитов и образование на их поверхности фибриновых волокон, способствующих консолидации первичной тромбоцитной пробки. Последующая дезинтеграция распространяется в глубь массы, чему способствует активация аутолитических ферментов, повышение проницаемости и растворение плазматических мембран. В результате создаются условия для повышенного проникновения из сыворотки крови в тромбоциты кальция, активации в них Са2+-зависимой АТФазы, дальнейшего сдвига соотношения АТФ/АДФ в сторону увеличения АДФ и, как следствие, дальнейшего и прогрессирующего усиления адгезии и агрегации. С момента распада тромбоцитов и выхода тромбоцитарных факторов свертывания крови в окружающую среду начинается следующий этап тромбоза – плазматическая фаза (фаза коагуляции крови). Физико-химическая и биохимическая сущность этой фазы заключается в нескольких последовательных превращениях по типу профермент-фермент. Некоторые из этих превращений имеют истинную ферментативную природу. На первом этапе фазы коагуляции крови происходит активация тромбопластина ткани и крови с переводом их в активный внешний и внутренний тромбопластин. Внешний тромбопластин образуется при взаимодействии тканевых и плазменных компонентов системы свертывания крови. Кровяной, или внутренний, тромбопластин (фактор 3 тромбоцитов) образуется из тромбоцитного протромбопластина при взаимодействии факторов свертывания плазмы. Время образования тканевого тромбопластина составляет несколько секунд, в то время как для образования кровяного тромбопластина требуются минуты. На втором этапе образуется активный тромбин. Под действием протеолитического фермента тромбопластина происходит отщепление пептидов с обоих концов белковой молекулы протромбина с образованием тромбина – высокоспецифичного протеолитического фермента. На третьем этапе под влиянием тромбина осуществляется превращение фибриногена в фибрин с образованием сгустка. В последующем межмолекулярные водородные связи в фибрине-полимере становятся еще более прочными под действием фибринстабилизирующего фактора плазмы крови. Фибрин в виде рыхло или компактно расположенных нитей представляет собой основную массу тромба. В ячейках образованной сети располагаются клетки крови (агрегированные тромбоциты, скопления лейкоцитов и эритроцитов). На заключительном этапе свертывания крови под действием тромбастенина (ретрактозима), который выделяется из интактных тромбоцитов, наступает сокращение (по типу сокращения актомиозина) фибриновых волокон и волоконец, обнаруженных в тромбоцитах с помощью электронного микроскопа. Происходит сжатие (ретракция) и уплотнение сгустка. Ретракция – лабильный процесс, нарушающийся при воздействии на тромбоциты химических (соли ртути, кобальта, меди, фтора, формальдегид, эфир, хлороформ) и физических (нагревание свыше 57°С, замораживание, воздействие ультразвука) факторов. При этом наблюдается полное подавление ретракции. Для нормального течения ретракции необходимо наличие ионов кальция, глюкозы, АТФ, физиологическое течение гликолиза, определенные соотношения между концентрацией тромбина и фибриногена, а также фибриногена и тромбоцитов. В принципе приведенная динамика процесса тромбообразования, в частности, обязательное наличие клеточной и плазматической фаз, характерна для большинства видов животных. Последствия тромбоза могут быть различными. Учитывая его значение как кровоостанавливающего механизма при острой травме, сопровождающейся кровотечением, тромбоз следует рассматривать с общебиологических позиций как приспособительное явление. В то же время тромбообразование при различных заболеваниях (атеросклероз, облитерирующий эндартериит, сахарный диабет и др.) может сопровождаться тяжелыми осложнениями, вызванными острым нарушением кровообращения в зоне тромбированного сосуда (ишемия при тромбозе артерий, застой крови при тромбозе вен). Развитие некроза (инфаркта, гангрены) в бассейне тромбированного, лишенного коллатералей сосуда – конечный этап тромбоза. Особенно велика роль тромбоза венечных артерий в развитии инфаркта миокарда. Исходом тромбоза могут быть асептическое (ферментативное, аутолитическое) расплавление, организация (рассасывание с замещением соединительной тканью), реканализация, септическое (гнойное) расплавление. Последнее особенно опасно, так как способствует септикопиемии и образованию множественных абсцессов в различных органах. Эмболия Эмболия (от греч. emballein – бросить внутрь) – закупорка сосудов телами (эмболами), приносимыми током крови или лимфы. В зависимости от характера эмболов различают эмболию экзогенную (воздушную, газовую, плотными инородными телами, бактериальную, паразитарную) и эндогенную, вызванную тромбом, жиром, различными тканями, околоплодными водами, газом (при кессонной болезни). По локализации различают эмболию большого, малого круга кровообращения и системы воротной вены. Во всех этих случаях движение эмболов обычно осуществляется в соответствии с естественным поступательным движением крови. Отсюда следует, что источником эмболии большого круга кровообращения являются патологические процессы в легочных венах, полостях левой половины сердца, артериях большого круга кровообращения; малого – патологические изменения в венах большого круга кровообращения и правой половине сердца. К возникновению эмболии воротной вены ведут патологические изменения в бассейне воротной вены. Исключением является ретроградная эмболия, когда движение эмбола подчиняется не гемодинамическим законам, а силе тяжести самого эмбола. Такая эмболия развивается в крупных венозных стволах при замедлении кровотока и уменьшении присасывающего действия грудной клетки. Различают также парадоксальную эмболию, которая наблюдается при незаращении межпредсердной или межжелудочковой перегородки, в результате чего эмболы из вен большого круга кровообращения и правой половины сердца переходят в левую, минуя малый круг (рис. 10.4).  Эмболия экзогенного происхождения. Воздушная эмболия возникает при ранении крупных вен (яремной, подключичной, синусов твердой мозговой оболочки), которые слабо спадаются и давление в которых близко к нулю или отрицательное. Данное обстоятельство может служить причиной воздушной эмболии и при осуществлении врачебных манипуляций – при инфузии растворов в указанные сосуды. В результате в поврежденные вены засасывается воздух, особенно, на высоте вдоха, с последующей эмболией сосудов малого круга кровообращения. Те же условия создаются при ранении легкого или деструктивных процессах в нем, а также при наложении пневмоторакса. В таких случаях, однако, наступает эмболия сосудов большого круга кровообращения. К аналогичным последствиям приводит поступление большого количества воздуха из легких в кровь при воздействии на человека взрывной ударной волны (воздушной, водяной), а также при "взрывной декомпрессии" и быстром подъеме на большую высоту. Возникающее при этом резкое расширение легочных альвеол, разрыв их стенки и поступление воздуха в капиллярную сеть приводят к неизбежной эмболии сосудов большого круга кровообращения. Чувствительность различных животных и человека к воздушной эмболии неодинакова. Кролик погибает от внутривенного введения 2 – 3 мл воздуха, в то время как собаки переносят введение воздуха в объёме 50–70 мл/кг. Человек в этом отношении занимает промежуточное положение. При анаэробной (газовой) гангрене возможна также газовая эмболия. Эмболия эндогенного происхождения. Источником тромбоэмболии является частица оторвавшегося тромба. Отрыв тромба считается признаком его неполноценности ("больной" тромб). В большинстве случаев "больные" тромбы образуются в венах большого круга кровообращения (вены нижних конечностей, таза, печени), чем и объясняется большая частота тромбоэмболии малого круга. Воспалительные изменения в клапанах легочного ствола и правом предсердно-желудочковом клапане, являющиеся основой тромбоэндокардита, нередко сопровождаются тромбоэмболией легочной артерии. Только в том случае, когда тромбы образуются в левой половине сердца (при эндокардите, аневризме) или в артериях (при атеросклерозе), наступает эмболия сосудов большого круга кровообращения. Причиной неполноценности тромба, отрыва его частиц и тромбоэмболии являются асептическое или гнойное расплавление его, нарушение фазы ретракции тромбообразования, а также свертывания крови. Жировая эмболия возникает при попадании в кровоток капель жира, чаще всего эндогенного происхождения. Причиной попадания жировых капель в кровоток является повреждение (размозжение, сильное сотрясение) костного мозга, подкожной или тазовой клетчатки и жировых скоплений, жирной печени. С возрастом в связи с замещением красного костного мозга трубчатых костей желтым и увеличением в нем содержания жиров с низкой температурой плавления опасность жировой эмболии возрастает. Поскольку источник эмболии располагается преимущественно в бассейне вен большого круга кровообращения, жировая эмболия возможна прежде всего в сосудах малого круга кровообращения. Лишь в дальнейшем возможно проникновение жировых капель через легочные капилляры (или артерио-венозные анастомозы малого круга) в левую половину сердца и артерии большого круга кровообращения. Количество жира, вызывающее смертельную жировую эмболию, колеблется у различных животных в пределах 0,9 – 3 см3/кг. Тканевая эмболия наблюдается при травме, когда возможен занос обрывков различных тканей организма, особенно богатых водой (костный мозг, мышцы, головной мозг, печень, трофобласт), в систему циркуляции крови, прежде всего малого круга кровообращения. Отрыв кашицеподобных жировых масс атером в атеросклеротически измененной артериальной стенке и попадание их в кровоток сопровождается эмболией артерий большого круга кровообращения. Особое значение имеет эмболия сосудов клетками злокачественных опухолей, поскольку является основным механизмом образования метастазов. Эмболия околоплодными водами возникает при попадании . околоплодных вод во время родов в поврежденные сосуды матки на участке отделившейся плаценты. В артериолах и капиллярах легких задерживаются плотные частицы амниотической жидкости (меконий, vernix caseosa), что сопровождается клиническим проявлением эмболии малого круга кровообращения. Такую разновидность эмболии от тканевой отличает повышение активности фибринолитической системы крови, резкое снижение содержания фибриногена в крови (гипо- и афибриногенемия), нарушение свертывания крови (вторичное) и длительно не прекращающееся кровотечение из матки. Газовая эмболия является основным патогенетическим звеном состояния декомпрессии, в частности кессонной болезни. Перепад атмосферного давления от повышенного к нормальному (у рабочих кессонов и водолазов) или от нормального к резко пониженному (при быстром подъеме на высоту или во время дегерметизации кабины высотного летательного аппарата) приводит к понижению растворимости газов (азота, углекислого газа, кислорода) в тканях и крови и закупорке пузырьками этих газов (в первую очередь азота) капилляров, расположенных главным образом в бассейне большого круга кровообращения. Клинические проявления эмболии определяются ее локализацией (малый или большой круг кровообращения), особенностями ангиоархитектоники, в частности, состоянием коллатерального кровообращения и его нейрогуморальной регуляции, размером и составом эмболов, их общей массой, скоростью попадания в кровоток, реактивностью организма. Эмболия малого круга кровообращения. Наиболее важным функциональным сдвигом при эмболии сосудов малого круга кровообращения является резкое снижение артериального давления в большом круге кровообращения и повышение давления в малом круге (рис. 10.5). Существует несколько гипотез, объясняющих механизмы гипотензивного эффекта при эмболии легочной артерии. Некоторые исследователи связывают резкое снижение артериального давления с уменьшением минутного объема крови, вызванным механической закупоркой легочной артерии и правожелудочковой недостаточностью сердца. Дальнейшие исследования, однако, показали, что механическое закрытие даже большей части сосудов легких еще не вызывает таких нарушений кровообращения, как при эмболии. Большое распространение получило мнение, согласно которому острое снижение артериального давления рассматривается как рефлекторная гипотензия (разгрузочный рефлекс Швигка-Парина). Считают, что депрессорный рефлекс при этом вызывается раздражением рецепторов, расположенных в русле легочной артерии. Как показали А. Б. Фохт и В. К. Линдеман (1903), ваготомия, а также введение атропина животным ослабляют степень депрессорной реакции, что подтверждает ее рефлекторный механизм. Определенное значение в снижении артериального давления при эмболии легочной артерии придают ослаблению функции сердца вследствие гипоксии миокарда, что является результатом увеличения нагрузки на правую половину сердца и резкого снижения артериального давления. Обязательным гемодинамическим эффектом эмболии сосудов малого круга кровообращения является повышение артериального давления в легочной артерии и резкое увеличение градиента давления на участке легочная артерия – капилляры, что рассматривается как результат рефлекторного спазма легочных сосудов. К такому же эффекту – раздражению рецепторов легочных сосудов и последующему спазму – может привести повышение давления в артериолах легких, механическое раздражение сосудов эмболами, уменьшение кровотока в сосуде ниже эмбола, выделение в месте закупорки веществ (серотонин, гистамин), обладающих свойством вызывать сокращение неисчерченных мышечных волокон сосудов. В связи с отмеченными гемодинамическими нарушениями резко повышается центральное венозное давление, развивается синдром острого легочного сердца (синдром острой правожелудочковой недостаточности сердца), который часто является причиной смерти. Нарушение гемодинамики в малом и большом круге кровообращения при эмболии легочной артерии в сочетании с генерализованным бронхиолоспазмом ведет к изменениям вентиляционно-перфузионного соотношения в легких и, как следствие, ко вторичным изменениям газового состава крови – увеличению напряжения СО2, уменьшению напряжения О2 (см. раздел XX – "Патологическая физиология внешнего дыхания"). В качестве приспособительной реакции, направленной на нормализацию газового состава крови, развивается одышка. Считают, что нарушение внешнего дыхания при эмболии легочной артерии является рефлекторной реакцией, возникающей как с рецепторного поля малого круга кровообращения, так и вследствие раздражения рефлексогенных зон большого круга кровообращения кровью с пониженным содержанием кислорода. Экспериментально показано, что перерезкой блуждающих нервов можно значительно ослабить степень наблюдаемых нарушений дыхания. Эмболия большого круга кровообращения. Как уже было сказано, в основе эмболии сосудов большого круга кровообращения чаще всего лежат патологические процессы в левой половине сердца, сопровождающиеся образованием на его внутренней поверхности тромбов (тромбоэндокардит, инфаркт миокарда), тромбообразование в артериях большого круга кровообращения с последующей тромбоэмболией, газовая или жировая эмболия. Местом частой локализации эмболов являются венечные, средняя мозговая, внутренняя сонная, почечная, селезеночная, брыжеечная артерии. При прочих равных условиях локализация эмболов определяется углом отхождения бокового сосуда, его диаметром, интенсивностью кровенаполнения органа. Большой угол отхождения боковых ветвей по отношению к вышерасположенному отрезку сосуда, сравнительно большой их диаметр, гиперемия являются факторами, предрасполагающими к той или иной локализации эмболов. При газовой эмболии, сопровождающей кессонную болезнь или "взрывную декомпрессию", предрасполагающим моментом к локализации эмболов в сосудах мозга и подкожной клетчатке является хорошая растворимость азота в богатых липоидами тканях. Тяжесть клинической картины в каждом конкретном случае определяется преимущественно взаимосвязью двух факторов – рефлекторного спазма сосудов и степени развития коллатералей. Рефлекторный . спазм может возникать не только в близлежащих сосудах, но и в отдаленных, усугубляя течение патологического процесса. В этом случае к местным патофизиологическим изменениям часто присоединяются общие, от которых нередко и погибают больные. С другой стороны, условия коллатерального кровообращения в бассейне сосуда, закупоренного эмболом, и в прилегающих тканях являются фактором, предотвращающим такие грозные и часто необратимые последствия эмболии, как омертвение соответствующего участка ткани. Эмболия воротной вены. Эмболия воротной вены, хотя и встречается значительно реже, чем эмболия малого и большого круга кровообращения, привлекает внимание в первую очередь характерным клиническим симптомокомплексом и чрезвычайно тяжелыми гемодинамическими нарушениями. Благодаря большой вместимости портального русла закупорка эмболом главного ствола воротной вены или основных его разветвлений приводит к увеличению кровенаполнения органов брюшной . полости (желудка, кишок, селезенки) и развитию синдрома портальной гипертензии (повышение давления крови в системе воротной вены с 8 – 10 до 40- 60 см вод. ст). При этом, как следствие, развиваются характерная клиническая триада (асцит, расширение поверхностных вен передней стенки живота, увеличение селезенки) и ряд общих изменений, вызванных нарушением кровообращения (уменьшение притока крови к сердцу, ударного и минутного объема крови, снижение артериального давления), дыхания (одышка, затем резкое урежение дыхания, апноэ) и функции нервной системы (потеря сознания, паралич дыхания). В основе этих общих нарушений лежит преимущественно уменьшение массы циркулирующей крови, вызванное скоплением (около 90%) ее в портальном русле. Такие нарушения гемодинамики часто являются непосредственной причиной смерти больных. В то же время между асцитом, расширением поверхностных вен передней стенки живота и спленомегалией, с одной стороны, и степенью портальной гипертензии, с другой, нет прямой зависимости. Иногда при высоком уровне портального давления указанные симптомы отсутствуют и, наоборот, в некоторых случаях они появляются даже при незначительном повышении давления в системе воротной вены. Это наводит на мысль, что в развитии указанных клинических проявлений портальной гипертензии, в частности асцита, кроме повышения давления большую роль играют и другие факторы, такие как нарушение обменных функций печени, задержка натрия и последующая задержка воды в организме, вызванная гиперпродукцией альдостерона корой надпочечных желез, вазопрессина нейросекреторными клетками супраоптического и паравентрикулярных ядер гипоталамуса; снижение коллоидоосмотического давления плазмы крови, обусловленное гипопротеинемией; повышение проницаемости стенки капилляров портального русла. |