зайко. Н. Н. Зайко Патологическая физиология Введение Предмет и задачи патологической физиологии Патологическая физиология есть наука, изучающая жизнедеятельность больного организма. Программа
 Скачать 7.32 Mb. Скачать 7.32 Mb.
|

|
Нарушение обмена витаминов при заболеваниях печени заключается в: 1. уменьшении всасывания жирорастворимых витаминов (ретинола, эргокальциферола, токоферолов, филлохинонов) в результате нарушения желчевыделительной функции печени (непоступление желчных кислот в кишечник); 2. нарушении синтеза витаминов и образования их биологически активных форм (ретинола из каротина, пиридоксальфосфата – активной формы витамина В6 и др.); 3. нарушении депонирования витаминов (цианокобаламина, фолиевой кислоты, никотиновой кислоты и др.) и их выведения из организма. В связи с этим различные патологические процессы в печени (вирусный гепатит, подпеченочная желтуха, гепатозы) могут сопровождаться развитием гиповитаминозов. Нарушение обмена гормонов и биологически активных веществ при патологии печени проявляется в изменении: 1. синтеза гормонов (в гепатоцитах из фенилаланина образуется тирозин – предшественник тироксина, трийодтиронина, катехоламинов), их транспортных белков (транскортина, связывающего 90 % гликокортикоидов); 2. инактивации гормонов (конъюгации стероидных гормонов с глюкуроновой и серной кислотой; ферментативного окисления катехоламинов под влиянием аминооксидаз, расщепления инсулина инсулиназой); 3. инактивации биологически активных веществ (окислительного дезаминирования серотонина и гистамина). Поражение печени патологическим процессом, при котором нарушается инактивация таких гормонов, как тироксин, инсулин, кортикостероиды, андрогены, эстрогены, ведет к сдвигам их содержания в крови и развитию эндокринной патологии. Уменьшение дезаминирования БАВ при патологии печени может усугубить клинические проявления аллергии. Нарушение обмена микроэлементов при заболеваниях печени связано с изменением: 1. депонирования в ней железа, меди, цинка, молибдена, марганца и др. (железа в форме ферритина и гемосидерина, кобальта – в виде цианокобаламина); 2. синтеза транспортных белков микроэлементов (например, трансферрина, церулоплазмина); 3. экскреции их с желчью. 1 Холестаз – нарушение оттока желчи с накоплением ее компонентов в печени и крови в результате снижения экскреторной функции гепатоцитов (первичный холестаз) или механического препятствия оттоку желчи в желчных протоках (вторичный холестаз). Нарушение защитной функции печени Поражение печени патологическим процессом сопровождается нарушением защитной (барьерной) функции печени, что проявляется в снижении фагоцитарной активности купферовских клеток (звездчатых эндотелиоцитов) и других макрофагальных элементов и антитоксической (обезвреживающей) функции. При заболеваниях печени (цирроз, гепатит, опухоль)угнетение фагоцитарной активности может быть обусловлено не только недостаточным кровоснабжением (гипоксией) и структурной деформацией органа, но и блокадой купферовских клеток образующимися в самой печени продуктами распада клеток и метаболитами. При этом уменьшается способность печеночных макрофагов элиминировать путем фагоцитоза из крови жировые капли, эритроциты, микроорганизмы, их токсины, особенно поступающие с портальным кровотоком из кишечника, что приводит к возникновению токсемии с разнообразными проявлениями (лейкоцитоз, лихорадка, гемолиз эритроцитов, почечная недостаточность, эрозии кишечника и др.). Наличие портокавальных анастомозов усугубляет течениетоксемического синдрома, который иногда протекает кактоксический шок в связи с поступлением токсических веществ из кишечника в системный кровоток на фоне выключенной фагоцитарной функции печени. Кроме того, при ослаблении фагоцитоза как неспецифической защитной реакции снижается устойчивость организма к инфекционным факторам. В то же время повышается частота развития аллергических (аутоаллергических) процессов как в самой печени, так и в других органах и системах, что обусловлено нарушением захвата из крови и разрушения макрофагами печени антигенов и иммунных комплексов (в норме в звездчатых ретикулоэндотелиоцитах расщепляется 95% веществ с антигенными свойствами). Понижение антитоксической функции печени связано с нарушением ее метаболической функции – синтеза мочевины (обезвреживание токсического аммиака), окисления (ароматических углеводородов), восстановления (нитробензола в парааминофенол), ацетилирования (сульфаниламидных препаратов), гидролиза (алкалоидов, сердечных гликозидов), конъюгации (образования парных соединений с глюкуроновой кислотой, гликоколом, цистеином, таурином – для связывания непрямого билирубина, скатола, фенола, индола и др.). Кроме того, при патологии печени нарушается еще один путь детоксикации – превращение водонерастворимых (аполярных) веществ в растворимые (полярные) соединения, которые могут быть выведены из организма с желчью и мочой. К ослаблению антитоксической функции печени приводит повреждение гепатоцитов как локусов обезвреживания, уменьшение активности ферментов, катализирующих реакции детоксикации, и дефицит энергии. Нарушение антитоксической функции печени при ее поражении может обусловить повышение чувствительности организма к различным лекарственным средствам – хинину, морфину, барбитуратам, наперстянке и др. Это связано с тем, что при уменьшении их расщепления в печени токсичность этих веществ для организма увеличивается, вызывая отравление. Кроме того, в процессе метаболических превращений токсических соединений в гепатоцитах могут образоваться еще более токсические вещества (синтез гепатотоксических веществ – метаболитов ряда медикаментов, например изониазида; образование канцерогенных веществ). Нарушение экскреторной функции печени при затруднении выделения желчи также может привести к накоплению токсических веществ в организме. Выключение антитоксической функции печени приводит к развитию гепатоцеребрального синдрома (печеночной энцефалопатии) и наиболее тяжелой формы клинического проявления печеночной недостаточности – печеночной комы. Гепатоцеребральный синдром, характеризующийся нарушениями психики, сознания и двигательными расстройствами (дрожание, атаксия, ригидность мышц), может перейти в печеночную кому. Печеночная кома – синдром, обусловленный токсическим поражением центральной нервной системы с глубокими расстройствами ее функций (потеря сознания, отсутствие рефлексов, судороги, нарушение кровообращения, дыхания), возникающий вследствие печеночно-клеточной (массивный некроз гепатоцитов) и сосудистой (наличие портокавальных анастомозов) форм печеночной недостаточности. Классификация. По патогенезу выделяют три разновидности печеночной комы: 1. печеночно-клеточную; 2. портокавальную; 3. смешанную (при некрозе паренхимы печени и недостаточном функционировании портокавальных анастомозов у больных циррозом). В эксперименте печеночная кома воспроизводится при помощи методики деваскуляризации печени и прямой фистулы Экка ("мясное отравление" собак). Этиология. Причиной печеночной комы может быть любое заболевание печени, но чаще всего вирусный гепатит, токсическая дистрофия печени, цирроз, острое расстройство печеночного кровообращения, синдром портальной гипертензии. Патогенез. Главным звеном в патогенезе печеночной комы является увеличение в крови содержания церебротоксических веществ, поступающих из кишечника в печень и не обезвреживаемых в ней при нарушении антитоксической функции гепатоцитов или образующихся в печени при разрушении паренхимы. При наличии шунтов между воротной и полыми венами эти вещества поступают из кишечника в общий кровоток, минуя печень (при синдроме портальной гипертензии). К церебротоксическим веществам прежде всего относитсяаммиак, наибольшее количество которого образуется в кишечнике из азотсодержащих соединений под влиянием ферментов кишечной палочки и протея. При патологии печени аммиак не включается в ней в орнитиновый цикл и не превращается в мочевину, а соединяется с ?-кетоглутаровой кислотой (образуется глутаминовая кислота и глутамин). Выключение ?-кетоглутаровой кислоты из цикла трикарбоновых кислот влечет за собой снижение продукции АТФ и угнетение АТФ-зависимых реакций. Вследствие снижения энергетического обмена в нейронах нарушается реполяризация нервных клеток и их функции. Кроме аммиака, церебротоксическим действием обладают и другие вещества, образующиеся в кишечнике: белковые метаболиты (фенолы, индол, скатол, амины, производные метионина), низкомолекулярные жирные кислоты(масляная, капроновая, валериановая), производныепировиноградной и молочной кислот (ацетоин, бутиленгликоль). Так, жирные кислоты, взаимодействуя с липидами мембран нервных клеток, тормозят передачу возбуждения в ганглиях. Возникающее при печеночной коме нарушение аминокислотного обмена и дисбаланс в их содержании в крови (увеличение фенилаланина, триптофана, тирозина, метионина и уменьшение валина, лейцина, изолейцина) может привести к синтезу ложных нейромедиаторов (октопамина, ?-фенилэтиламина), которые вытесняют такие медиаторы, как норадреналин и допамин, прерывают передачу возбуждения в синапсах в центральной нервной системе, что также обусловливает неврологическую симптоматику при печеночной коме. Нарушение церебральных функций при коме связано и с непосредственным действием церебротоксических веществ на трансмембранный потенциал в результате увеличения активности Na+, К+-зависимой АТФазы и в связи с этим повышения проницаемости мембран нейронов. Важнейшими звеньями в патогенезе печеночной комы являются гипогликемия (следствие снижения гликонеогенеза в патологически измененных гепатоцитах), усиливающая дефицит энергии в клетках мозга; гипоксия(нарушение транспорта кислорода в связи с блокадой дыхательной поверхности эритроцитов токсическими веществами и гемодинамическими расстройствами); сдвиги водно-электролитного обмена и кислотно-основного состояния. Изменение водно-электролитного обмена при печеночной коме проявляется в развитии гипокалиемии, возникающей как следствие вторичного гиперальдостеронизма при снижении метаболизма альдостерона в печени (увеличивается реабсорбция натрия в канальцах нефрона и выведение калия с мочой). При потере внеклеточного калия и на фоне повышения проницаемости клеточных мембран происходит выход калия из клеток, в том числе мозга, по градиенту концентрации в межклеточное пространство, а ионы натрия и водорода поступают в клетки. Развивается внутриклеточный ацидози гипокалиемический внеклеточный алкалоз, что способствует проникновению аммиака в нервные клетки и усилению его церебротоксического действия. В то же время при печеночно-клеточной коме нередко отмечаетсяметаболический ацидоз не только в клетках печени, но и мозга вследствие накопления пировиноградной, молочной, трикарбоновых, жирных кислот, в связи с чем повышается проницаемость клеточных мембран, возрастает поступление церебротоксических веществ и воды в клетки мозга (отек). Нарушение желчевыделения. Этиология. Причины нарушения прохождения желчи по желчевыводящим путям в двенадцатиперстную кишку могут быть следующие: 1. механическое препятствие оттоку желчи при сдавлении желчевыводящих путей извне (опухолью головки поджелудочной железы, воспаленной тканью, рубцом) или их закупорке (камнем, гельминтами, густой желчью); 2. нарушение иннервации желчевыводящих путей – гипер- и гипокинетические дискинезии (например, уменьшение желчевыделения при спазме сфинктера шейки желчного пузыря); 3. изменение гуморальной регуляции желчевыделения (желчевыделение усиливается при гиперпродукции секретина, холецистокинина, мотилина). Патогенез нарушения желчевыделения рассматривается ниже (см. "Подпеченочная желтуха"). Нарушение желчеобразовательной и желчевыделительной (экскреторной) функции печени Печеночные клетки секретируют желчь, в состав которой входят желчные кислоты, желчные пигменты, холестерин, фосфолипиды, жирные кислоты, муцин, вода и другие вещества. Печень участвует в образовании, метаболизме и экскреции желчных пигментов (рис. 22.2). В звездчатых эндотелиоцитах печени, в макрофагах костного мозга, селезенки из гемоглобина разрушенных эритроцитов образуется вердоглобин, из него после отщепления атома железа и глобина – биливердин, который превращается, восстанавливаясь, в билирубин. В крови билирубин соединяется преимущественно с альбумином, и этот комплекс называется свободным, неконъюгированным, непрямым билирубином (непрямой, так как окраску с диазореактивом Эрлиха дает только после осаждения белка). Он нерастворим в воде, в норме составляет 75 % от общего билирубина крови (6,8 – 20,5 мкмоль/л по методу Ендрашика и соавт.), нетоксичен, не проникает в мозг и, следовательно, не может вызвать билирубиновую энцефалопатию. Свободный билирубин, не включенный в билирубин-альбуминовый комплекс, легко проходит через гематоэнцефалический барьер и, взаимодействуя с фосфолипидами мембран нейронов, поступает в клетки центральной нервной системы и может их повредить. Однако в норме концентрация свободного билирубина в крови настолько мала, а способность альбумина его связывать столь высока, что он не оказывает токсического действия. 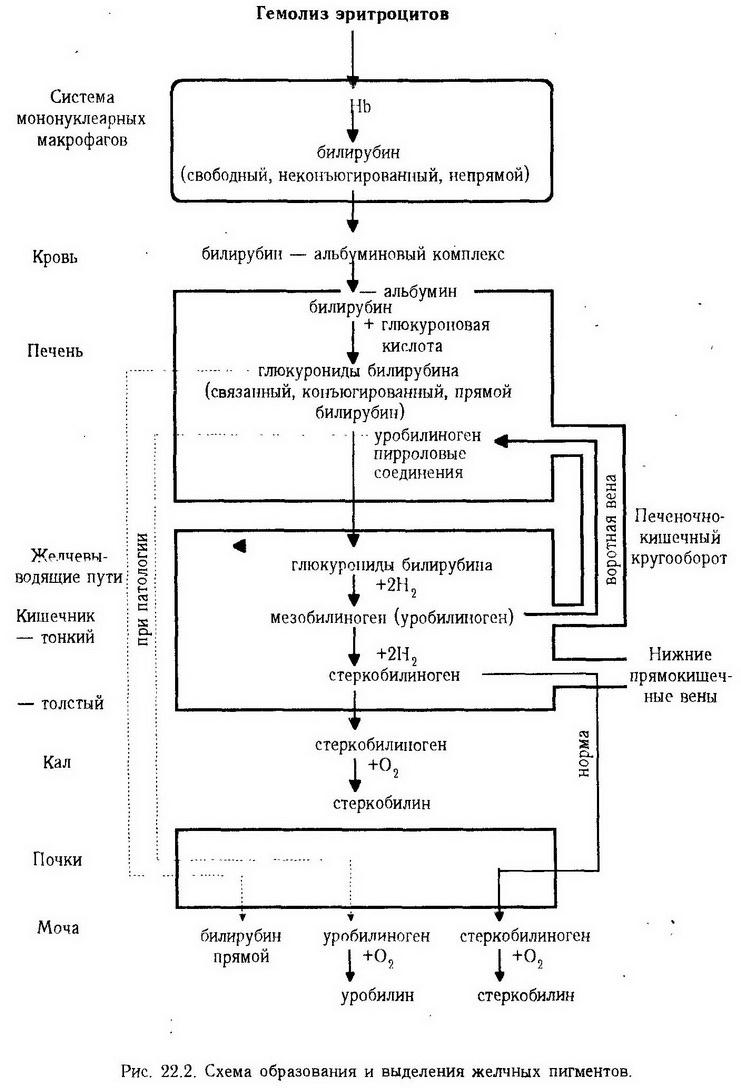 У своего васкулярного полюса гепатоциты осуществляют захват из крови неконъюгированного билирубина, от которого в цитоплазматической мембране отделяется альбумин. В переносе билирубина через мембрану клетки, а затем из цитоплазмы в мембрану эндоплазматического ретикулума участвуют белки Y (лигандин) и Z (глутатионтрансферазы). В мембране эндоплазматического ретикулума гепатоцита билирубин конъюгируется с уридиндифосфоглюкуроновой кислотой (УДФГК) под влиянием фермента УДФ-глюкуронилтрансферазы и образуется моноглюкуронид билирубина (МГБ), который у билиарного полюса гепатоцита переходит через гепатоканаликулярную мембрану в желчь по градиенту концентрации, создаваемому желчными кислотами (энергозависимый процесс). В желчных канальцах из двух молекул МГБ образуется диглюкуронид билирубина (ДГБ) при участии билирубин-глюкуронидтрансферазы. Таким образом, в желчи содержится конъюгированный билирубин (в основном ДГБ), растворимый в воде, так называемый связанный, или прямой, билирубин (дает прямую реакцию с диазореактивом). Во внепеченочных ходах, желчном пузыре и тонком кишечнике (главным образом) от ДГБ под действием ферментов кишечной микрофлоры отщепляется глюкуроновая кислота (деконъюгация), свободный билирубин проникает в кровь, а оставшийся ДГБ восстанавливается доуробилиногена (мезобилиногена), часть которого всасывается через кишечную стенку в кровь и из воротной вены поступает в печень (печеночно-кишечный кругооборот), где разрушается до пиррольных соединений. В связи с тем что уробилиноген в норме в общий кровоток не попадает, он отсутствует и в моче. Большая часть уробилиногена в толстом кишечнике восстанавливается до стеркобилиногена, выделяемого с калом в виде окисленной формы – стеркобилина. Незначительное количество стеркобилиногена, всосавшись в кровь в нижнем отделе толстого кишечника, через нижние геморроидальные вены поступает в систему нижней полой вены и выводится с мочой, в которой в норме содержатся следы стеркобилиногена. Однако в клинической практике его принято называть уробилиногеном или же уробилиновыми телами (общий термин для выделяемых с мочой продуктов обмена билирубина). Нарушение желчеобразования проявляется в увеличении или уменьшении секреции желчи, как правило, с одновременным изменением ее состава. Этиология. Причинами увеличения или уменьшения желчеобразования могут быть следующие: 1. изменение нейрогуморальной регуляции (например, увеличение секреции желчи при усилении тонуса блуждающего нерва или повышении инкреции секретина и гастрина); 2. алиментарные факторы, некоторые лекарственные растения и препараты (жиры, яичный желток, настой кукурузных рылец, сорбит, дефицит белка); 3. экзогенные и эндогенные факторы, нарушающие энергетический обмен в организме, в том числе и в печени, не пораженной патологическим процессом (гипоксия, перегревание, гипотермия, отравление цианидами); 4. поражение печени и желчевыводящих путей (гепатит, гепатоз, холецистит), приводящее к нарушению секреторной функции гепатоцитов вследствие их дистрофии и деструкции и изменению реабсорбции компонентов желчи; 5. снижение активности кишечной микрофлоры, уменьшающее печеночно-кишечный кругооборот компонентов желчи и, следовательно, их концентрацию в желчи (при патологических процессах в тонкой кишке, под влиянием антибиотиков); 6. нарушение образования и обмена билирубина и желчных кислот и изменение их содержания в желчи. Патогенез. К механизмам, обусловливающим количественные и качественные нарушения желчеобразования, относятся: 1. изменение секреторной активности гепатоцитов; 2. нарушение реабсорбции компонентов желчи в желчевыводящих путях и кишках (печеночно-кишечный кругооборот); 3. сдвиги в транс- и интерцеллюлярной фильтрации некоторых веществ из крови в печеночные капиллярные сосуды. В связи с тем что секреция желчи и реабсорбция ее компонентов являются энергозависимыми процессами, в основе их нарушения лежат энергетические сдвиги в тканях (нарушение активности ферментов биологического окисления, сопряженности тканевого дыхания и окислительного фосфорилирования и др.). Изменение фильтрация обусловлено колебаниями осмотического градиента и состоянием проницаемости мембран. Нарушение печеночно-кишечного кругооборота некоторых компонентов желчи происходит при изменении ферментативной активности микрофлоры кишок, которая обеспечивает биохимическое превращение желчных кислот и связанного билирубина (образование их производных, деконъюгирование, разрушение) и последующую реабсорбцию в тонкой кишке, возвращение с кровью в печень и повторную экскрецию с желчью. Количество реабсорбированных в кишках желчных кислот определяет интенсивность их биосинтеза в печени и влияет на холатохолестериновый индекс желчи, играющий важную роль в ее литогенных свойствах, т. е. способности желчи к образованию камней. Патофизиологические синдромы, обусловленные нарушением желчеобразования и желчевыделения Нарушения желчеобразования и желчевыделения проявляются в виде следующих синдромов: желтухи, холемии, ахолии, дисхолии. Желтуха (icterus) – синдром, возникающий при увеличении содержания в крови билирубина и характеризующийся желтой окраской кожи, слизистых оболочек, склер в результате отложения в них желчных пигментов при нарушении желчеобразования и желчевыделения. Классификация. В зависимости от первичной локализации патологического процесса, приводящего к развитию желтухи, и механизма возникновения выделяют такие виды желтухи: 1. надпеченочную, вызванную повышенной продукцией билирубина, главным образом в связи с усилением распада эритроцитов (гемолитическая желтуха) и реже при нарушении плазменного транспорта билирубина; 2. печеночную желтуху, обусловленную нарушением захвата, конъюгации и экскреции билирубина гепатоцитами вследствие их повреждения при различных патологических процессах, а также приобретенных и наследственных дефектах структуры гепатоцитов и ферментов, участвующих в метаболизме и транспорте билирубина в клетках печени; 3. подпеченочную желтуху (механическую), возникающую при затруднении оттока желчи по внепеченочным желчевыводящим путям. Надпеченочная желтуха. К этой группе относятся: 1. гемолитическая желтуха, которая развивается в результате повышенного распада эритроцитов; 2. шунтовая гипербилирубинемия – при возрастании образования так называемого шунтового билирубина из гемоглобина незрелых форм эритроцитов (например, нормобластов костного мозга в результате неэффективного эритропоэза при В12-дефицитной анемии) или же из гема таких протеидов, как миоглобин, цитохромы, каталаза, при обширных гематомах, инфарктах; 3. желтуха при нарушении плазменного транспорта билирубина – при разрыве связи между билирубином и альбумином некоторыми медикаментами или же нарушении образования билирубин-альбуминового комплекса вследствие резкого уменьшения содержания альбумина в крови. Этиология. Причины возникновения гемолитической желтухи – это те этиологические факторы, которые приводят к развитию гемолиза эритроцитов и гемолитической анемии (см. раздел XVIII – "Гемолитическая анемия"). Патогенез. При усиленном гемолизе эритроцитов в звездчатых эндотелиоцитах печени, макрофагах селезенки, костного мозга образуется столь большое количество свободного (непрямого, неконъюгированного) билирубина, что гепатоциты печени оказываются не в состоянии полностью извлечь его из крови и связать с уридиндифосфоглюкуроновой кислотой (относительная печеночная недостаточность). Кроме того, гемолитические яды часто являются гепатотоксическими веществами, а поражение гепатоцитов затрудняет метаболизм и транспорт билирубина в них. В крови увеличивается содержание неконъюгированного билирубина (непрямая гипербилирубинемия), который не выводится с мочой из-за своей связи с альбумином. Может возрасти уровень конъюгированного (прямого) билирубина, что обусловлено его обратной диффузией в кровь после того, как способность гепатоцита экскретировать связанный билирубин в желчь оказалась исчерпанной. При очень высокой непрямой гипербилирубинемии (260 – 550 мкмоль/л), когда не весь свободный билирубин включается в билирубин-альбуминовый комплекс, развивается так называемая ядерная желтуха (окрашивание ядер головного мозга) с поражением ядер центральной нервной системы и неврологической симптоматикой (энцефалопатия), что особенно характерна для гемолитической болезни (анемии) новорожденных при резус- несовместимости эритроцитов матери и плода. Токсическое действие свободного билирубина на нервную систему может проявиться и при незначительном повышении билирубина в крови, но наличии гипоальбуминемии, повышении проницаемости гематоэнцефалического барьера, мембран нервных клеток (при нарушении обмена липидов, гипоксии). При гемолитической желтухе в печени, желчевыводящих путях и кишечнике синтезируется избыточное количество глюкуронидов билирубина, уробилиногена, стеркобилиногена (гиперхолия – увеличенная экскреция желчи в кишечник), что приводит к повышенному выделению стеркобилина и уробилина с калом и мочой. Однако при этом отсутствуют холемический синдром (желчные кислоты в кровь не поступают) и расстройство кишечного пищеварения (нет ахолического синдрома, как при других желтухах). К гемолитической желтухе могут присоединиться печеночная желтуха, если одновременно с гемолизом будут поражены гепатоциты, и механическая желтуха вследствие закупорки желчевыводящих путей желчными тромбами и камнями из билирубина, холестерина и кальция. Печеночная желтуха. Этиология. Причиной возникновения печеночной желтухи является прежде всего действие этиологических факторов, вызывающих повреждение гепатоцитов (инфекция, токсические, в том числе лекарственные, вещества, внутрипеченочный холестаз), а также наследственный дефект захвата, конъюгации и выведения билирубина из гепатоцита. Патогенез. Выделяют следующие патогенетические разновидности печеночных желтух: 1. Печеночная желтуха вследствие нарушения захвата билирубина гепатоцитом может возникнуть: • в результате уменьшения в печеночной клетке содержания белков Y (лигандина) и Z, обеспечивающих перенос билирубина через цитоплазматическую мембрану из крови в клетку (при белковом голодании); • из-за конкурентного торможения захвата билирубина (рентгеноконтрастными веществами, некоторыми медикаментами, например антигельминтными препаратами); • вследствие генетически детерминированного нарушения структуры мембраны васкулярного полюса гепатоцитов (это ведет к изменению проницаемости мембраны), отщепления в ней свободного билирубина от связи с альбумином и переноса билирубина в гепатоцит (при наследственном синдроме Жильбера – Мейленграхта). При этом уже вторично не происходит конъюгация билирубина и возникает непрямая гипербилирубинемия (увеличивается количество свободного билирубина). В моче и кале снижается содержание стеркобилина из-за нарушения образования глюкуронидов билирубина в гепатоцитах и, следовательно, их производных в желчных канальцах и кишечнике. 2. Печеночная желтуха вследствие нарушения конъюгации билирубина с уридиндифосфоглюкуроновой кислотой в мембране эндоплазматического ретикулума возникает при снижении активности УДФ-глюкуронилтрансферазы, катализирующей этот процесс. Такой механизм развития печеночной желтухи отмечается при физиологической желтухе новорожденных, у недоношенных детей, при вскармливании грудным молоком матери с высоким содержанием прегнандиола (эстрогены подавляют активность УДФ-глюкуронилтрансферазы, конкурируя с билирубином за связь с ней), применении ряда лекарственных препаратов (викасол), гипотиреозе, а также наследственном дефиците фермента (синдром Криглера – Найяра, синдром Жильбера). Отсутствие или понижение активности УДФ-глюкуронилтрансферазы обусловливает нарушение образования связанного билирубина, количество которого в желчи уменьшается, что приводит к снижению выделения стеркобилина с калом и мочой. В то же время в крови возрастает концентрация неконъюгированного билирубина -непрямая гипербилирубинемия. Высокий уровень свободного билирубина при синдроме Криглера–Найяра вызывает тяжелую энцефалопатию благодаря развитию ядерной желтухи. 3. Печеночная желтуха вследствие нарушения экскреции билирубина из гепатоцита в желчевыводящие пути развивается при изменении проницаемости билиарной части цитоплазматической мембраны печеночной клетки, цитолизе гепатоцитов, разрыве желчных канальцев, сгущении желчи и закупорке внутрипеченочных путей (внутрипеченочный холестаз). Изолированное нарушение выведения конъюгированного билирубина имеет место при наследственных синдромах Дубина – Джонсона (с выраженной пигментацией печени в результате накопления в ней субстратов типа меланина как последствие сниженной экскреторной функции гепатоцита) и Ротора, когда в крови увеличивается содержание связанного билирубина – прямая гипербилирубинемия, отмечаетсябилирубинурия и в то же время пониженное выделение стеркобилина с калом и мочой. Значительно чаще уменьшение выделения билирубина в той или иной мере сочетается с нарушением его захвата, внутриклеточного транспорта, конъюгации гепатоцитом. Таков механизм возникновения печеночной желтухи при повреждении клеток печени (гепатоцеллюлярная желтуха) и внутрипеченочном холестазе (холестатическая желтуха), что наблюдается при вирусных, инфекционных, токсических (в том числе медикаментозных) гепатитах, обменных гепатозах, циррозе печени (например, первичном билиарном циррозе), диффузной инфильтрации печени при лейкозах, гемохроматозе. При повреждении печеночных клеток возникает сообщение между желчными путями, кровеносными и лимфатическими сосудами, через которое желчь поступает в кровь и частично в желчевыводящие пути. Отек перипортального пространства также может способствовать обратному всасыванию желчи из желчных ходов в кровь. Набухшие клетки сдавливают желчные протоки, создавая механическое затруднение оттоку желчи. Метаболизм и функции печеночных клеток нарушаются. При гепатоцеллюлярной и холестатической разновидности печеночной желтухи резко снижается экскреция конъюгированного билирубина в желчь, и он поступает из патологически измененных гепатоцитов в кровь, возникаетпрямая гипербилирубинемия. В то же время в крови повышается уровень свободного билирубина – непрямая гипербилирубинемия, что связано со снижением таких функций гепатоцита, как захват, внутриклеточный транспорт свободного билирубина и его связывание в глюкурониды. Попадание в кровь вместе с желчью желчных кислот обусловливает развитие холемического синдрома. Уменьшение поступления желчи в кишечник (гипохолия,ахолия) ведет к понижению образования метаболитов билирубина и их выделения с калом и мочой (следы стеркобилина), а также появлению симптомов ахолического синдрома. Насыщенный желтый цвет мочи объясняется повышенным содержанием в ней прямого билирубина (билирубинурия)и уробилина, который недостаточно разрушается в печени после поступления в нее благодаря печеночно-кишечному кругообороту, попадает в общий кровоток и выводится почками (уробилинурия). Повреждение печеночных клеток воспалительно-дистрофическим процессом при гепатоцеллюлярной и холестатической формах печеночной желтухи сопровождается развитием не только экскреторной, но и печеночно-клеточной разновидности печеночной недостаточности с нарушением всех функций печени, в том числе метаболической и защитной. При этом нередко понижается свертывание крови. Подпеченочная желтуха (механическая, обтурационная). Этиология желтухи изложена в подразделе "Нарушение желчевыделения". Патогенез. Механическое препятствие оттоку желчи приводит к застою (внепеченочный вторичный холестаз) и повышению давления желчи выше 2,7 кПа (270 мм вод. ст.), расширению и разрыву желчных капилляров и поступлению желчи прямо в кровь или через лимфатические пути. Появление желчи в крови обусловливает прямую гипербилирубинемию (увеличивается содержание конъюгированного билирубина), гиперхолестеринемию, развитие холемического синдрома в связи с циркуляцией в крови желчных кислот, билирубинурию (отсюда темная окраска мочи – "цвета пива") и наличие желчных кислот в моче. Непоступление желчи в кишечник из-за механического препятствия в желчевыводящих путях приводит к тому, что не образуется и, следовательно, не выделяется стеркобилин с калом (обесцвеченный, ахоличный кал) и мочой. Таков же механизм развития ахолического синдрома, наиболее выраженного при механической желтухе при полной обтурации желчевыводящих путей. Холемический синдром, наблюдаемый при механической и печеночной желтухе (гепатоцеллюлярная и холестатическая желтуха), возникает при попадании желчных кислот в кровь. Он характеризуется брадикардией и снижением артериального давления при действии желчных кислот на рецепторы и центр блуждающего нерва, на синусовый узел сердца и кровеносные сосуды. Токсическое действие желчных кислот на центральную нервную систему проявляется в виде общей астении, раздражительности, сменяющейся депрессией, сонливости днем и бессонницы ночью, головной боли и повышенной утомляемости. Раздражение чувствительных нервных окончаний кожи желчными кислотами приводит к кожному зуду. Увеличение содержания желчных кислот в крови может вызвать гемолиз эритроцитов, лейкоцитолиз, снижение свертывания крови, повышение проницаемости мембран и развитие воспалительного процесса на месте контакта с тканями (печеночный некроз, перитонит, острый панкреатит). Ахолический синдром обусловлен непоступлением желчи в кишечник при обтурации желчевыводящих путей или нарушении экскреторной функции гепатоцита (при механической и печеночной желтухе). При этом наблюдается расстройство кишечного пищеварения. Вследствие отсутствия в кишках желчных кислот не активируется липаза, не эмульгируются жиры, не образуются растворимые комплексы желчных кислот с жирными кислотами, в связи с чем 60 – 70 % жиров не переваривается, не всасывается и удаляется из организма вместе с калом (стеаторея). Нарушение всасывания жирорастворимых витаминов (ретинола, токоферола, филлохинона) приводит к развитиюавитаминозов. Без филлохинона (витамина K1) не образуется протромбин, снижается свертывание крови, что обусловливает повышенную кровоточивость Отсутствие желчных кислот приводит к нарушению моторики кишечника: ослабляются тонус и перистальтика кишок, появляется запор. Однако последний нередко сменяется поносом в связи с усилением гнилостных и бродильных процессов в кишках и снижением бактерицидных свойств желчи. Кал обесцвечен, так как при ахолии не образуется стеркобилин, который исчезает и из мочи. Дисхолия, при которой желчь приобретает литогенные свойства, обусловливает образование желчных камней в желчном пузыре и желчных протоках и развитие желчно-каменной болезни. Этиология. Причины дисхолии разнообразны: воспалительные процессы, дискинезия желчного пузыря, желчных протоков, заболевания пищевого канала, избыточное содержание холестерина в пище, нарушение обмена веществ (особенно холестеринового, билирубинового). Патогенез. Одним из основных механизмов возникновения литогенной желчи является снижение холатохолестеринового и лецитинохолестеринового индексов (отношения желчных кислот и лецитина к холестерину желчи). Это может быть вызвано уменьшением печеночно-кишечного кругооборота желчных кислот при патологии кишок и изменении в них микрофлоры, угнетением синтеза желчных кислот в печени (при понижении активности 7?-гидроксилазы), ускорением их всасывания слизистой оболочкой воспаленного желчного пузыря, уменьшением содержания лецитина и увеличением синтеза холестерина. При уменьшении концентрации желчных кислот и лецитина, обеспечивающих взвешенное состояние холестерина, холестерин выпадает в осадок и дает начало образованию холестериновых камней. Инфекция, застой желчи также способствуют процессу камнеобразования, так как сопровождаются изменением свойств желчи – сдвигом рН в кислую сторону, снижением растворимости солей, выпадением их в осадок, коагуляцией белков из распадающихся клеток. Помимо холестериновых, образуются пигментные (при гемолизе эритроцитов), известковые и сложные камни (например, холестериново-пигментно-известковые). Камни обусловливают нарушение желчевыделения и развитие механической желтухи. Нарушение гемодинамики, эритропоэза и свертывания крови при патологии печени Нарушение гемодинамической функции печени проявляется в изменении объема депонированной крови (в печени депонируется в норме около 800 мл крови) и сосудистого тонуса. Увеличение или уменьшение депонирования крови в печеночных венах играет важную роль в изменении объема циркулирующей крови при патологии сердечно-сосудистой системы, водно-электролитного обмена и др. Выход крови из сосудов печени в общий кровоток является компенсаторной реакцией при кровопотере, высотной болезни. Нарушение гемодинамической функции печени может привести к развитию артериальной гипотензии и синдрома портальной гипертензии. В основе возникновения печеночного гипотензивного синдрома лежит уменьшение в пораженной патологическим процессом печени синтеза ангиотензиногена (т. е. отсутствует субстрат для образования ангиотензина при действии ренина) и повышенная продукция ферритина, вызывающего расширение сосудов. Кроме того, как было сказано выше, гипотензивным влиянием обладают желчные кислоты (при холемическом синдроме, наблюдаемом при паренхиматозной и механической желтухе). Повышение сосудистого сопротивления в системе воротной вены приводит к развитию синдрома портальной гипертензии, для которого характерно увеличение давления в этой системе. В зависимости от места возникновения препятствия оттоку крови выделяют три разновидности портальной гипертензии: надпеченочную (при тромбозе, эмболии, сдавлении нижней полой вены и печеночных вен, правожелудочковой недостаточности сердца, перикардите), внутрипеченочную (при циррозе, метастазах опухолей в печени), подпеченочную (тромбоз, сдавление воротной вены). Главным звеном в патогенезе портальной гипертензии является застой крови в системе воротной вены. Компенсаторное развитие коллатерального кровообращения частично обеспечивает отток крови через анастомозы воротной вены с полыми, печеночной артерией и через новообразованные сосуды с печеночными венами. Застой крови в органах брюшной полости ведет к изменению их функций, нарушению общей гемодинамики и асциту – скоплению жидкости в брюшной полости. Развитие портокавальных анастомозов является не только проявлением компенсации, но и причиной возникновения кровотечений из варикозно расширенных коллатералей, а также печеночной энцефалопатии и комы. Нарушение эритропоэза при поражении печени патологическим процессом связано с уменьшением депонирования в ней таких необходимых для кроветворения факторов, как цианокобаламин, фолиевая кислота, железо, что приводит к развитию анемии. Патологические изменения гепатоцитов могут обусловитьснижение свертывания крови вследствие уменьшения синтеза протромбина, факторов V, VII, IX, X и фибриногена. Кроме того, при печеночной и механической желтухе возникает гиповитаминоз К как следствие гипо- и ахолии и нарушения всасывания жиров. При недостатке этого витамина в организме снижается образование протромбина и других факторов свертывания крови. Все сказанное выше объясняет частоту геморрагического диатеза при заболеваниях печени. |