зайко. Н. Н. Зайко Патологическая физиология Введение Предмет и задачи патологической физиологии Патологическая физиология есть наука, изучающая жизнедеятельность больного организма. Программа
 Скачать 7.32 Mb. Скачать 7.32 Mb.
|

|
Возникающая при нарушении реабсорбции белка тубулярная протеинурияможет быть двух типов: 1. связанная с нарушением реабсорбции белка; 2. обусловленная поступлением в мочу белковых молекул из разрушенных клеток канальцев. Первый тип тубулярной протеинурии наблюдается при отравлении кадмием, фенацетином, при гипоксии, ожогах, при гипервитаминозе D, трансплантации почек, септицемии, при острой недостаточности почек, при синдроме Фанкони (см. ниже), дисметаболических поражениях почек. Характеризуется невысоким содержанием в моче альбуминов и наличием преимущественно низкомолекулярных белков (до 70 000) (селективная протеинурия). Поскольку в нормальных условиях эпителий канальцев осуществляет практически полную реабсорбцию указанных белков, то их появление в моче считается признаком избирательного (селективного) нарушения механизма реабсорбции. При тяжелых формах поражения канальцевого аппарата, характеризующихся наличием грубых дистрофических изменений клеток канальцев, в моче появляются белки с большой относительной молекулярной массой (свыше 70 000). Такую протеинурию рассматривают как неселективную. В механизме ее возникновения имеет значение нарушение способности эпителия канальцев подвергать ферментативному расщеплению крупномолекулярные белки, попавшие в мочу через поврежденный клубочковый фильтр[1]. При втором типе тубулярной протеинурии в моче могут появляться гиалиновые, эпителиальные восковидные и зернистые цилиндры. Белковую основу цилиндров составляет уропротеин Тамма – Хорсфалла, продуцируемый эпителием извитых канальцев. Зернистые и восковидные цилиндры, как правило, являются признаком органического поражения почечной паренхимы. Нарушение реабсорбции глюкозы Нарушение реабсорбции глюкозы проявляется в видеглюкозурии – выделения глюкозы с мочой. Глюкозурия внепочечного происхождения возникает во всех случаях гипергликемии, превышающей "почечный порог", – 8,8 – 10 ммоль/л (160 – 180 мг %). Почечная глюкозурия проявляется на фоне нормального или даже пониженного уровня глюкозы в крови. Наблюдается при хронических заболеваниях почек, интоксикации свинцом, ртутью, ураном, а также как наследственная аномалия, передающаяся по доминантному типу. Основным механизмом почечной глюкозурии является снижение активности (приобретенные формы) или генетически обусловленный дефицит ферментов (гексокиназа, глюкозо-6-фосфатаза), обеспечивающих канальцевую реабсорбцию глюкозы. В эксперименте почечную глюкозурию воспроизводят с помощью флоридзина – ингибитора фосфорилирования в клетках канальцев нефронов. Выраженные формы почечной глюкозурии сопровождаются полиурией, возникающей по механизму осмотического диуреза. Нарушения реабсорбции неорганического фосфата и кальция. Наследственный фосфатный почечный диабет. Проявляется значительным повышением суточной экскреции фосфатов и вследствие этого снижением уровня их в плазме крови; нормальным или слегка пониженным уровнем кальция в крови, повышенной кальциурией; деминерализацией костей; различными формами рахита (у детей) и остеомаляции (у взрослых), резистентными к лечению большими дозами витамина D. В большинстве случаев заболевание имеет наследственный характер. Фосфатный почечный диабет характеризуется первичным поражением проксимальной части канальцев нефронов в отношении их способности реабсорбировать фосфаты или повышением чувствительности канальцев нефронов к паратирину (псевдогиперпаратиреоидизм). Наследственная остеодистрофия (псевдогипопаратиреоидизм) характеризуется такими же изменениями в крови, как и истинный гипопаратиреоз, – гипокальциемией, гиперфосфатемией. Однако применение паратирина не устраняет эти изменения и не повышает экскрецию фосфатов с мочой вследствие резистентности канальцев нефронов к действию гормонов. Считают, что такая резистентность обусловлена отсутствием или дефектом соответствующих рецепторов к паратирину в канальцах нефронов. Нарушение реабсорбции аминокислот Проявляется аминоацидурией – повышенным выделением свободных аминокислот с мочой (норма около 1,1 г/сут). Возможно нарушение транспортного механизма одной или нескольких аминокислот. Аминоацидурия характеризуется ренальной или экстраренальной тубулярной дисфункцией, может быть генетически обусловленной (первичная) или приобретенной (вторичная). Ренальная аминоацидурия развивается на фоне нормального или пониженного (в зависимости от продолжительности и выраженности аминоацидурии) содержания аминокислот в плазме крови. Основным механизмом возникновения ренальной аминоацидурии является наследственно обусловленный дефицит ферментов или коферментов, участвующих в транспорте аминокислот. Описаны наследственные тубулярные синдромы, характеризующиеся цистинурией, глицинурией и др. Большинство из них сопровождается симптомами мочекаменной болезни и ее осложнений. Аминоацидурия экстраренального происхожденияв некоторых случаях является следствием наследственных заболеваний обмена веществ и вторичного поражения канальцев в связи с увеличенным содержанием в крови аминокислот (фенилкетонурия, лейциноз, гиперпролинемия, гиперглицинемия, цистиноз, оксалоз и др.). Нарушение реабсорбции аминокислот вызвано недостаточностью ферментов из-за "перегрузки" транспортного механизма или токсического действия на почки продуктов промежуточного обмена. При этом могут наблюдаться нарушения ацидо-, аммониогенеза, водно-электролитного обмена, протеинурия, глюкозурия, кальцийурия. Нередкими являются кальцификация различных структур почек (нефрокальциноз), развитие почечнокаменной болезни. Аминоацидурия может развиваться также как следствие усиленного катаболизма белков, нарушения промежуточного обмена аминокислот в условиях гипоксии, голодания, дефицита никотиновой к ислоты, витаминов группы В, при ожоговой болезни, при обширном инфаркте миокарда, тяжелых поражениях печени. Ее возникновение связано с гипераминоацидемией и "перегрузкой" транспортного механизма канальцев нефронов. Комбинированные тубулопатии. Анатомическая близость и определенная биохимическая общность (энергозависимость процесса) механизмов, обеспечивающих реабсорбцию глюкозы, фосфатов, аминокислот, способствуют появлению сочетанных нарушений указанных канальцевых функций и соответствующих тубулярных синдромов. Наиболее известные из них – глюкофосфатный диабет, глюкозурия с аминоацидурией, сочетанное нарушение реабсорбций аминокислот и гидрокарбонатов, фосфатов и некоторых аминокислот. Самым сложным по проявлению и тяжелым по клиническому течению является синдром Фанкони. Для этого генетически обусловленного состояния характерно одновременное нарушение реабсорбции глюкозы, фосфатов, гидрокарбонатов, аминокислот, а также развитие канальцевого ацидоза (вследствие потери гидрокарбонатов) и гипокалиемии. В некоторых случаях наблюдаются нарушение концентрационной способности почек и обезвоживание организма вследствие осмотического диуреза. Как правило, это состояние сопровождается гипофосфатемическим рахитом с нормокальциемией, резистентным к лечению витаминами группы D. Клиническую картину, близкую к таковой при синдроме Фанкони, наблюдали при интоксикации солями тяжелых металлов (ртуть, свинец, уран). Нарушения канальцевой секреции. Ведущим тубулярным синдромом, в основе которого лежит нарушение канальцевой секреции, является канальцевый ацидоз. Основной механизм развития канальцевого ацидоза – торможение аммонио- и ацидогенеза и секреции Н+ в канальцах нефронов, что затрудняет взаимозависимую реабсорбцию Na+ и гидрокарбонатов и выведение из организма кислых продуктов в виде титруемых кислот. Конкретные причины и механизмы торможения секреции ионов водорода при канальцевом ацидозе не установлены. Полагают, что он возникает при нарушении ферментативных процессов цикла Кребса и недостаточности канальцевой глутаминазы, участвующей в образовании аммиака из глутамина. Чаще всего канальцевый ацидоз является следствиемтубулоинтерстициального синдрома – особой формы поражения почек, для которой характерно развитие атрофии эпителия канальцев нефронов, в основном дистальной части их, в сочетании с выраженным склерозом стромы и нарушением основных канальцевых функций в ответ на различные токсические, метаболические, физические (ионизирующая радиация) и инфекционные воздействия. Нарушение секреции мочевой кислоты сопровождается повышением концентрации в крови мочевой кислоты и ее солей (гиперурикемия) и развитием почечной формы подагры. Соответствующий дефект является наследственно обусловленным и передается по доминантному типу. Нарушение секреции чужеродных веществ (лекарственных – антибиотики, красящих – фенолрот, йодсодержащих контрастных препаратов). Наблюдается при поражениях почек с выраженным тубулоинтерстициальным синдромом. Задержка в крови в связи с нарушением секреции некоторых чужеродных веществ, в частности пенициллина и продуктов его превращения, может сопровождаться токсическими проявлениями. ____________________ [1] Неселективная протеинурия свидетельствует о повреждении всего нефрона – повышении проницаемости клубочковой мембраны и повреждении эпителия канальца. Почечная недостаточность Под недостаточностью почек понимают такое изменение почечных функций, которое вызывает нарушение гомеостаза. Различают острую и хроническую почечная недостаточность. Каждая из указанных форм недостаточности почек в свою очередь делится на тубулярную (см. выше) и тотальную, обусловленную сочетанным нарушением функций клубочков и канальцев. Основным показателем, который определяет сочетанный или изолированный характер нарушений почечных функций, является степень уменьшения массы действующих нефронов (МДН). Независимо от этиологии заболевания при уменьшении массы действующих нефронов более чем в два раза наблюдается нарушение всех почечных процессов (клубочковой фильтрации, проксимальной реабсорбции глюкозы, канальцевого транспорта натрия, осмотического концентрирования и. разведения мочи и др.). При умеренной степени уменьшения МДН наблюдаются изолированные нарушения почечных функций. Острая почечная недостаточность Острая почечная недостаточность характеризуется остро возникающим нарушением постоянства внутренней среды организма из-за значительного и быстрого снижения скорости клубочковой фильтрации (в норме 120 мл/мин, при олиго- и анурии – 1 – 10 мл/мин). Этиология. Острая почечная недостаточность (ОНП) связана с действием 3 групп факторов: преренальных, ренальных и постренальных. Преренальные факторы ОНП: 1. кровопотеря, ожоги, неукротимая рвота, профузные поносы, использование диуретиков, вследствие чего резко уменьшается объем внутрисосудистой и внеклеточной жидкости; 2. сосудистые формы шока (септический, анафилактический), коллапс, сопровождающиеся увеличением емкости сосудистого русла и падением артериального давления; 3. острая (инфаркт миокарда, эмболия легочной артерии) и хроническая недостаточность сердца. Ренальные факторы ОНП: 1. местные нарушения кровообращения в почках (тромбоз, эмболия почечной артерии, тромбоз почечных вен; ишемия, обусловленная преренальными факторами; синдром внутрисосудистого свертывания крови); 2. острые заболевания почек воспалительной природы – острый интерстициальный нефрит, острый гломерулонефрит, васкулит; 3. нефротоксические влияния (антибиотики, соли тяжелых металлов, органические растворители, рентгеноконтрастные вещества, токсикоз беременных, диабетическая кома, интоксикация грибным и змеиным ядом; анаэробная инфекция, сепсис и перитонит, печеночная недостаточность; 4. повреждающее действие пигментов (гемоглобина – при массивном внутрисосудистом гемолизе, миоглобина – при массивном травматическом и нетравматическом рабдомиолизе). Постренальные факторы ОНП: 1. обструкция мочеточников (камни, опухоли, сгустки, некротические массы – изнутри; опухоли, увеличенные лимфатические узлы, спайки – извне); 2. задержка выделения мочи на уровне выхода из мочевого пузыря (аденома, опухоль простаты). Патогенез. Основным механизмом развития ОНП является временная ишемия почек, преимущественно коркового вещества, обусловленная гиповолемией, спазмом афферентных артериол, диссеминированным внутрисосудистым свертыванием крови с микротромбообразованием или непосредственным поражением почечных сосудов. Следствием этого является выраженное снижение фильтрационного давления и клубочковой фильтрации, выключение деятельности определенного количества нефронов. Если нарушение почечного кровотока непродолжительно, то ОНП является обратимым состоянием (функциональная фаза ОНП). Затяжная ишемия вызывает необратимые структурные изменения клубочков и канальцев, что соответствует структурной фазе ОНП. При действии нефротоксических факторов (токсических, инфекционных) наряду с нарушением кортикального кровотока важное значение приобретает прямое повреждение структур клубочков и канальцев. При этом скорость клубочковой фильтрации может уменьшаться и вторично – в связи с обструкцией просвета канальцев некротическими массами или в связи с утечкой фильтрата через стенку поврежденных канальцев в интерстиций. Повышение давления в капсуле Шумлянского – Боумена или в интерстиции приводит к падению эффективного фильтрационного давления. Допускается возможность вторичного снижения скорости клубочковой фильтрации по механизму клубочково-канальцевой обратной связи. В условиях повреждения клеток проксимальных канальцев нарушается реабсорбция Na+. Повышенная концентрация последнего в дистальных канальцах воспринимается macula densa, что приводит к активации ренин- ангиотензиновой системы, спазму афферентных артериол, уменьшению кровотока и скорости клубочковой фильтрации. При обструктивных состояниях мочевыводящих путей причиной уменьшения скорости клубочковой фильтрации является повышение давления в капсуле Шумлянского – Боумена (в ранние сроки ОНП – до 12 ч). Позже снижается и интенсивность почечного кровотока (под действием ангиотензина и тромбоксана А2). В клиническом течении ОНП выделяют четыре стадии: 1. начальную; 2. олигоанурии; 3. полиурии; 4. выздоровления. Наиболее характерные и выраженные нарушения наблюдаются в стадии олигоанурии. Наряду с резким снижением диуреза вплоть до полного его прекращения наблюдаются гиперазотемия, нарушение водно-электролитного гомеостаза и кислотно-основного равновесия. Основные клинические проявления этой стадии: отек головного мозга, интерстициальный отек легких, в целом клиническая картина водного отравления организма; тяжелые нарушения деятельности системы кровообращения – понижение сократительной функции сердца, нарушения ритма в виде экстрасистолии, брадикардии, блокады; гипотензия с последующим переходом в гипертензию, расстройство дыхания по типу Куссмауля (признак ацидоза), тяжелые расстройства функций нервной системы – головная боль, рвота, арефлексия, нарушение сознания, судороги, кома, прогрессирующая анемия и др. Все эти явления обусловлены сдвигами в состоянии гомеостаза. Большая часть больных, страдающих острой недостаточностью почек, погибает на высоте этой стадии. При более благоприятном течении заболевания, а главное, при проведении эффективных терапевтических мероприятий спустя 5 – 10 дней наступает переход в стадию восстановления диуреза и полиурии. Повышение клубочковой фильтрации обусловлено как восстановлением этого процесса в действующих нефронах (в начальном периоде полиурии), так и последующим (по истечении нескольких месяцев) увеличением МДН. Позднее восстанавливаются и другие почечные функции (способность к концентрированию мочи, аммонио- и ацидогенез и др.). Хроническая почечная недостаточность. Уремия. Этиология. Этиологическими факторами хронической недостаточности почек (ХНП) являются хронические прогрессирующие заболевания почек воспалительной (хронический гломерулонефрит, хронический пиелонефрит и др.), сосудистой (гипертоническая болезнь, стеноз почечной артерии) и метаболической (диабетический гломерулосклероз, амилоидоз, подагра) природы. Патогенез. ХНП развивается в результате одновременного или последовательного уменьшения массы действующих нефронов и соответственно снижения почечных функций. Начальные признаки ХНП появляются при снижении МДН до 50 – 30% исходного количества нефронов, клинически выраженная картина развивается при снижении МДН до 30 – 10 % и величины клубочковой фильтрации ниже 20%. Дальнейшее снижение МДН и клубочковой фильтрации (ниже 10% от нормы) приводит к развитию терминальной стадии недостаточности почек – уремии. Основные проявления ХНП обусловлены прежде всего азотемией вследствие понижения экскреции конечных продуктов азотистого обмена. Степень азотемии отражает степень уменьшения МДН. Обнаружено свыше 200 токсических веществ (уремических токсинов), накопление которых в крови при ХНП определяет интоксикацию организма и связанные с ней анорексию (отсутствие аппетита), диспепсические явления (рвота, понос), снижение массы тела, общую слабость, головную боль, апатию, нарушения вкуса, слуха, мучительный зуд, полиневрит, нарушение дыхания, прогрессирующую анемию, уремический перикардит, миокардит, плеврит, артрит, судороги, кому. В начальной стадии ХНП диурез сохранен и несколько усилен (полиурия), что обусловлено резким ограничением реабсорбции воды в дистальной части канальцев нефронов и собирательных трубочках, понижением концентрационной способности почек (гипо-, изостенурия). Олигурия характерна лишь для терминальной стадии ХНП – уремии. Нарушения осмотического и объемного гомеостаза, а также азотемия в значительной степени определяются снижением МДН. При полиурии возможны также гиповолемия, внутри- и внеклеточная дегидратация, гипонатриемия, более постоянны гипокалиемия, гипокальциемия, гипермагниемия. В олигоанурической (уремической) стадии ХНП наблюдаются гиперволемия, гипергидратация вне- и внутриклеточного пространства, клиническая картина водного отравления, проявляющаяся в виде отека головного мозга, легких, гипонатриемией, гиперкалиемией, гипокальциемией, с которой связывают развитие остеодистрофии и остеомаляции. Существенное значение имеют также нарушения кислотно-основного равновесия в виде ренального (азотемического) ацидоза. Общая характеристика основных синдромов и заболеваний почек Заболевания почек различной природы наблюдаются у 1,5 – 2% населения. Они составляют около 5,5 – 6% общей заболеваемости, характеризуются затяжным течением и высокой летальностью. В основе развития заболеваний почек лежат различные по характеру патологические процессы – воспаление, в том числе аллергической природы, типические расстройства местного кровообращения, дисметаболические нарушения, опухолевое перерождение и др. Многообразные этиологические факторы и патологические процессы довольно часто воспроизводят сходные морфологические, функциональные и клинические проявления изменений почечных и внепочечных функций. В их основе лежит сходство непосредственных механизмов, ответственных за повреждение почечных структур. Именно в этом заключается развитие "больших" клинических синдромов (острая и хроническая почечная недостаточность, нефротический синдром) при различных нозологических формах, разумеется, в различной степени выраженности. В то же время действие некоторых этиологических факторов при наличии варьирующего комплекса сопутствующих условий характеризуется выраженным полиморфизмом изменений в почках и, вероятно, различными особенностями их патогенеза. Примером может служить диабетическая нефропатия. Гломерулонефрит Гломерулонефрит является наиболее часто встречающимся двусторонним диффузным заболеванием почек воспалительной природы. Разнообразные изменения в клубочках нефронов представляют собой сочетание развивающихся интракапиллярно экссудативного и пролиферативного процессов. При тяжелом течении заболевания вследствие некротических изменений наблюдается полная деструкция клубочков. В патологический процесс вовлекается практически вся масса клубочков обеих почек. Различают острый и хронический (диффузный) гломерулонефрит. Острый (диффузный) гломерулонефрит. Экспериментальные модели. В 1901 г. В. К. Линдеман в лаборатории И. И. Мечникова наблюдал основные проявления нефрита у кролика при внутривенном введении ему нефротоксической сыворотки морской свинки, предварительно иммунизированной взвесью кроличьей почки. Используя аналогичную схему постановки опытов, японский ученый Masugi в 1933 г. воспроизвел клиническую картину нефрита у кроликов при введении им сыворотки крови уток, иммунизированных тканью почек кроликов. Другой вариант модели нефрита Masugi осуществлен по схеме крыса – кролик – крыса. В настоящее время в патогенезе экспериментального гломерулонефрита различают две фазы: гетерологичную, обусловленную фиксацией нефротоксических антител (IgG, IgM) на базальной мембране клубочков нефрона, иаутологичную, связанную с выработкой комплементфиксирующих антител на нефротоксический глобулин. Некоторые авторы допускают возможность и третьей, или аутоиммунной, фазы, обусловленной антителами к измененным сосудистым клубочкам. В 1909 г. русский хирург П. А. Герцен, пытаясь выяснить механизм возникновения "окопного" нефрита (нефрит военного времени), получил его экспериментальную модель у кролика путем охлаждения (замораживания хлорэтилом) почки. При этом в крови подопытных животных были обнаружены специфические противопочечные антитела. Особенностью данной модели было поражение не только поврежденной, но и интактной почки. Данная модель впервые навела на мысль о возможности аутоиммунной природы данного заболевания. Итальянскими учеными супругами Ковелти (1945) был получен гломерулонефрит у кроликов путем введения им в брюшную полость клеточной взвеси почечной ткани и культуры стрептококков. С достаточной надежностью острый гломерулонефрит воспроизводится путем введения животным чужеродного белка, сыворотки. R. W. Stablay (1962) воспроизвел гломерулонефрит у овец при иммунизации их базальными мембранами клубочков нефрона почки человека в полном адъюванте Фрейнда. W. Heymann и соавт. (1965) воспроизвели экспериментальный нефрит у крыс путем их активной иммунизации взвесью гомологичной или аутологичной почки с полным адъювантом Фрейнда. У новозеландских мышей линии NZB/BL спонтанно возникают серологические и морфологические признаки аутоиммунного гломерулонефрита и других аутоиммунных заболеваний: системной красной волчанки, аутоиммунной гемолитической анемии. На базальной мембране пораженных клубочков. обнаружен аутологичный у-глобулин, представляющий собой аутоантитела к почечным аутоантигенам. Появление заболевания регистрируется с 3 – 5-месячного возраста. Этиология. Острый гломерулонефрит возникает при (или после) какой-либо инфекции, чаще стрептококковой природы. Считается, что гемолитический стрептококк группы А (типа 4, 12) является специфическим "нефритогенным" штаммом. Определенную роль играют и другие инфекции, в том числе вирусные, паразитарные. Установлена этиологическая роль в возникновении гломерулонефрита охлаждения, диффузных поражений соединительных тканей (красная волчанка, ревматоидный артрит, узелковый периартериит), предшествующей вакцинации или использования с лечебной целью гетерологических сывороток, ожоговой болезни. Патогенез. Отсутствие инфекционного начала, например стрептококков, в почках при развитии в них диффузного воспалительного процесса, наличие латентного периода (1 – 3 нед) в течении постинфекционного гломерулонефрита, возникновение его при заболеваниях аутоиммунной природы, при сывороточной болезни и вакцинации, при экспериментальном моделировании с помощью иммунологического вмешательства, результаты серологических и иммунофлюоресцентных исследований позволяют предполагать аллергическую и аутоаллергическую природу данного заболевания. В соответствии с этим выделяют два основных механизма поражения клубочков (рис. 23.2). 1. Поражение базальной мембраны клубочков нефронов антителами против ее антигенов – нефротоксический гломерулонефрит (протекает быстро с прогрессирующим течением). Носителем антигенных свойств базальной мембраны является гликопротеид. 2. Развитие воспалительного процесса в клубочках вследствие фиксации на базальной мембране и интрамембранно иммунных комплексов -иммунокомплексный гломерулонефрит. 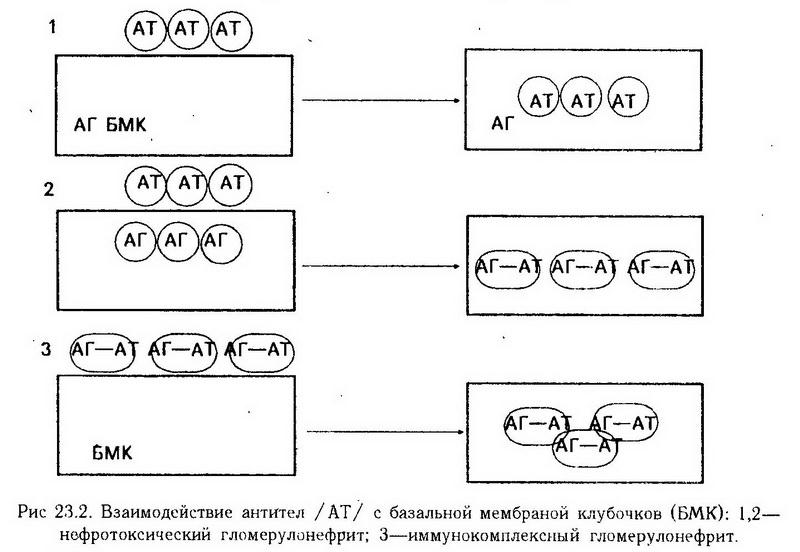 В качестве антигена при этом механизме служит экзогенный (инфекционного или неинфекционного происхождения) или эндогенный (тканевой белок, ДНК) антиген. Образующиеся антитела (IgG, IgM) непосредственно в сыворотке крови вступают во взаимодействие с указанными антигенами, затем в виде иммунных комплексов (антиген – антитело – комплемент) поступают в клубочки, откладываясь на их базальной мембране. Реализация повреждающего действия иммунных комплексов, как и нефротоксических антител, осуществляется путем индукции иммунного воспаления (см. раздел VII – "Аллергия"). К иммунокомплексный относятся гломерулонефриты, развивающиеся после стрептококковой инфекции, при системной красной волчанке, сывороточной болезни и др. Большинство случаев гломерулонефрита (не менее 80%) относятся к иммунокомплексный. Клинические и патофизиологические проявления острого гломерулонефрита отражают изменения почечных, в основном клубочковых, и внепочечных функций. Классическое течение заболевания характеризуется бурным началом, олигурией, протеинурией, гематурией, азотемией, артериальной гипертензией, отеками, которые развиваются вследствие задержки натрия, гипопротеинемии, гиперволемии, повышения проницаемости капиллярных сосудов; нарушениями со стороны центральной нервной системы. Хронический (диффузный) гломерулонефрит Представляет собой длительное прогрессирующее, диффузное двустороннее поражение почек воспалительной природы, неоднородное по происхождению, клиническим проявлениям и патогенезу. Этиология. Хронический гломерулонефрит может возникнуть как следствие острого, но чаще развивается первично. Выделяют следующие формы хронического гломерулонефрита: 1. инфекционного происхождения (постстрептококковый, при затяжном септическом эндокардите, малярии, сифилисе, туберкулезе и других, инфекциях); 2. неинфекционные (сывороточный, вакцинный, лекарственный, при отравлении различными ядами, травматический, при охлаждении, при тромбозе почечных вен); 3. при диффузных заболеваниях соединительной ткани (ревматоидный артрит, красная волчанка, геморрагический васкулит и др.); 4. особые (постэклампсический, радиационный, наследственный и др.). Патогенез. Общепризнанной является иммунологическая концепция развития хронического гломерулонефрита. Наряду с двумя основными механизмами, с которыми связывают развитие как острого, так и хронического гломерулонефрита, определенное значение имеет гиперчувствительность замедленного типа. Клинически в функционально-компенсированной фазе заболевания выделяют следующие формы. 1. Латентная (65% от всех больных хроническим гломерулонефритом) форма проявляется изолированным мочевым синдромом – умеренной протеинурией, гематурией. У части больных (20 – 25%) наблюдаются отеки и транзиторная гипертензия. 2. Гипертоническая (32% больных) форма характеризуется стойким повышением артериального давления. У 1/3 больных – отеки, у 2/3 – гематурия, у всех больных – протеинурия, у половины – цилиндрурия и лейкоцитурия. 3. Нефротическая (2 – 4% больных) форма, которую отличают отечный синдром (у 2/3 больных), выраженная протеинурия и цилиндрурия (у всех больных), характерные изменения в крови (гипопротеинемия, гиперлипидемия). 4. Смешанная, или нефротически-гипертоническая (2,4% больных) форма, для которой характерны отеки и гипертензия (у всех больных), те же, что и при нефротической форме, изменения в моче, однако в отличие от нее без характерных изменений в крови. Нарушение клубочково-канальцевого равновесия в функционально-компенсированной фазе хронического гломерулонефрита характеризуется преимущественными по сравнению с клубочковыми (несущественное уменьшение величины клубочковой фильтрации) изменениями тубулярной функции в форме изолированных или сочетанных синдромов (повышение проксимальной реабсорбции ионов натрия и воды, понижение экскреции "осмотически свободной" воды, понижение способности к концентрированию мочи, к секреции аммиака и ионов водорода. Масса действующих нефронов у большинства больных сохранена или незначительно уменьшена. В фазе декомпенсации в связи со значительными склеротическими изменениями ("сморщенная" почка) и понижением вследствие этого МДН хронический гломерулонефрит проявляет себя синдромом хронической недостаточности почек. Нефротический синдром Нефротический синдром включает в себя разнообразные болезненные состояния почек и других органов, для которых характерными являются выраженная протеинурия и гипопротеинемия, диспротеинемия, гиперлипидемия, отечный синдром. По этиологии, патогенезу, морфогенезу эти состояния заметно отличаются друг от друга. Этиология. Нефротический синдром по происхождению делят на первичный и вторичный. Первичный нефротический синдром не связан с каким-либо предшествующим заболеванием почек. Чаще всего в основе его возникновения имеется наличие генетически обусловленных дефектов обмена веществ (липоидный нефроз) или трансплацентарный перенос специфических противопочечных антител от матери к плоду (врожденный семейный нефроз). Вторичный нефротический синдром обусловлен некоторыми заболеваниями почек (гломерулонефрит) или других органов (нефропатия беременных, сахарный диабет, амилоидоз, красная волчанка, сывороточная болезнь, стафилококковый сепсис и др.). Наблюдается также при отравлении солями тяжелых металлов, при обширных ожогах, радиационном поражении, при отторжении почечного трансплантата, при применении некоторых лекарственных препаратов (сульфаниламиды, пенициллин, кортикостероиды), при нарушении кровоснабжения почек. Для экспериментального моделирования нефротического синдрома используют соли тяжелых металлов, пуромицин, а также иммунологические воздействия (например, введение суспензии из ткани гомологичной почки или противопочечной цитотоксической сыворотки). Патогенез. Большинство нефротических состояний обусловлено иммунологическими механизмами, преимущественно гиперчувствительностью замедленного типа. Источниками антигенов могут быть экзогенные факторы: бактериальные, вирусные, паразитарные, лекарственные, пищевые, соединения тяжелых металлов и др. В качестве эндогенных антигенов служат ДНК, денатурированные нуклеопротеиды, белки опухолевого происхождения, тиреоглобулин. Антитела, продуцируемые в ответ на указанные антигены, принадлежат преимущественно к классу IgM. Поражение клубочков почечных телец связывают с отложением на поверхности или в самой базальной мембране капиллярных сосудов амилоида, глико- и липопротеидов, фибриногена с активацией гуморальных и клеточных звеньев воспалительной реакции. Вследствие этого утрачивается структурная целостность базальной мембраны, изменяются ее состав и физико-химические свойства, резко повышается ее проницаемость для плазменных белков. Для тех форм нефротического синдрома, для которых иммунологические механизмы не доказаны, наиболее приемлемыми являются метаболический и физико-химический механизмы. Исходя из них, нефротическая протеинурия объясняется уменьшением постоянного электрического заряда стенки капиллярной сети, исчезновением из нее сиалопротеина, в норме тонким слоем покрывающего эндотелий и его отростки. В местах, где потеря анионов и сиалопротеинов является максимальной, скапливаются полиморфно-ядерные лейкоциты, лизосомальные ферменты которых оказывают непосредственное повреждающее действие на базальную мембрану капиллярных сосудов. Протеинурия в свою очередь обусловливает вторичные изменения канальцев нефронов и стромы почек, а также ряд общих изменений в организме: гипопротеинемию идиспротеинемию (гипоальбуминемия, гипер-?2-глобулинемия), возникновение отечного синдрома. Определенную роль в возникновении последнего, кроме гипопротеинемии и увеличения проницаемости мембран, играет вторичный гиперальдостеронизм, развивающийся вследствие гиповолемии (причиной является "утечка" жидкости в ткани), снижения почечного кровотока и повышенной продукции ренина. Свойственная нефротическому синдрому гиперлипидемияобусловлена преимущественно триглицеридами, холестерином и патогенетически связана с нарушениями белкового обмена и угнетением липолитической активности плазмы крови. Пиелонефрит Пиелонефрит представляет собой инфекционно-воспалительное заболевание слизистой оболочки мочевых путей и паренхимы почек (одновременное или последовательное) с преимущественным поражением интерстициальной ткани. Этиология и патогенез. Заболевание возникает в связи с занесением возбудителя инфекции в почки гематогенным путем или распространением его в восходящем направлении по мочевым путям. Возбудителями чаще всего являются кишечная палочка, кокки. Возникновению заболевания, переходу острого пиелонефрита в хронический способствуют различные условия, вызывающие застой мочи (сужение, закупорка мочеточников, аденома простаты), нарушение трофики мочевых путей, общие заболевания, снижающие реактивность организма (сахарный диабет, атеросклероз, ожирение, хроническая интоксикация и др.). Начинается пиелонефрит как острое заболевание, которое в большинстве случаев (исключая случаи полного выздоровления) через латентную, бедную симптомами фазу переходит в хроническую форму, заканчивающуюся сморщиванием и недостаточностью почек. Клиническое течение пиелонефрита характеризуется признаками тяжелого инфекционного процесса, проявляющегося выраженной интоксикацией (особенно в острой стадии); лихорадкой, лейкоцитозом; развитием артериальной гипертензии в 25 % случаев в начальной стадии и в 70 % – в далекозашедшей стадии; умеренно выраженным отечным синдромом и анемией; мочевым синдромом [полиурия, в поздней стадии – олигурия, поллакиурия – частые мочеиспускания; гипостенурия, в заключительной стадии – изостенурия; лейкоцитурия, гематурия, умеренная (5 – 10 г/л) протеинурия, цилиндрурия]. Нарушение клубочково-канальцевого равновесия характеризуется явным преобладанием канальцевых дисфункций над клубочковыми, особенно в начальных стадиях заболевания (своеобразная функциональная диссоциация). Об этом свидетельствует понижение способности почек к концентрированию мочи вследствие нарушения процесса реабсорбции жидкости, ранний и тяжелый канальцевый ацидоз, связанный с нарушением ацидо-и аммониогенеза,синдром потери солей, в основе которого имеется резкое понижение канальцевой реабсорбции ионов натрия, калия и кальция. Как следствие указанных расстройств могут развиваться опасные для жизни нарушения водно-электролитного обмена и кислотно-основного равновесия организма. Прогрессирование заболевания сопровождается нарастанием описанных нарушений, переходом тубулоинтерстициальной недостаточности в общую почечная недостаточность, снижением МДН и развитием хронической недостаточности почек. Последствия нарушений недиуретических функций почек Артериальная гипертензия. Развивается в результате уменьшения почечного кровотока, чаще всего при остром или хроническом поражении клубочков нефронов (очаговый и диффузный гломерулонефрит, хронический пиелонефрит, диабетический гломерулосклероз), при сужении или закрытии просвета почечных артерий и их ветвей вследствие аномалии развития, атеросклероза, тромбоза, эмболии, сдавления рубцом или опухолью. Механизмы возникновения "почечной" гипертензии изложены в разделе "Патологическая физиология системного кровообращения". Анемия. При заболеваниях почек, характеризующихся преимущественным поражением клубочков нефронов, сопровождающимся той или иной степенью нарушения их экскреторной функции (острый и хронический гломерулонефрит и др.), часто наблюдается анемия. Как правило, она является нормоцитарной, нормохромной, гипорегенераторной. Патогенетически возникновение такого рода анемии связывают главным образом с увеличением выработки ингибитора эритропоэза и (или) понижением продукции эритропоэтина на фоне увеличенной выработки ингибитора эритропоэза. Следствием уменьшения или выпадения эритропоэтической функции юкстагломерулярного аппарата почек является торможение синтеза ДНК в эритропоэтинчувствительных клетках костного мозга, нарушение их дифференцировки, угнетение пролиферации нормоцитов и выброса ретикулоцитов из костного мозга в кровь. Дополнительную роль в развитии "почечной" анемии играют угнетение деятельности костного мозга азотсодержащими веществами, гематурия, проявления геморрагического синдрома, дефицит железа, большая потеря трансферрина с мочой при сильно выраженной протеинурии, дефицит цианокобаламина. Нарушения коагуляции крови. Исследование свертывающей и антисвертывающей систем крови при заболеваниях почек [очаговый нефрит, острый и хронический (диффузный) гломерулонефрит] позволило установить, с одной стороны, гиперкоагуляцию крови и понижение активности фибринолиза – с другой. Лишь в терминальной стадии недостаточности почек развивается гипокоагуляция крови с клиническими проявлениями геморрагического синдрома. Основными причинами его развития являются дефицит некоторых факторов (тромбопластин, проконвертин) свертывания крови, тромбоцитопения, повышение уровня плазменных антикоагулянтов (гепарина), активация фибринолиза. |