Ответы к экзамену По биохимии 1 структура и функции белков 3 ферменты 10 нуклеиновые кислоты и нуклеотиды 21
 Скачать 29.52 Mb. Скачать 29.52 Mb.
|

|
Железо. В цельной крови железо содержится в основном в эритроцитах (около 18,5 ммоль/л), в плазме концентрация его составляет в среднем 0,02 ммоль/л. Ежедневно в процессе распада гемоглобина эритроцитов в селезенке и печени освобождается около 25 мг железа и столько же потребляется при синтезе гемоглобина в клетках кроветворных тканей. В костном мозге (основная эритропоэтическая ткань человека) имеется лабильный запас железа, превышающий в 5 раз суточную потребность в железе. Значительно больше запас железа в печени и селезенке. Повышение содержания железа в плазме крови наблюдается при ослаблении синтеза гемоглобина или усиленном распаде эритроцитов. При анемии различного происхождения потребность в железе и всасывание его в кишечнике резко возрастают. Известно, что в двенадцатиперстной кишке железо всасывается в форме двухвалентного железа. В клетках слизистой оболочки кишечника железо соединяется с белком апоферритином и образуется ферритин. Предполагают, что количество поступающего из кишечника в кровь железа зависит от содержания апоферритина в стенках кишечника. Дальнейший транспорт железа из кишечника в кроветворные органы осуществляется в форме комплекса с белком плазмы крови трансферрином. Железо в этом комплексе трехвалентное. В костном мозге, печени и селезенке железо депонируется в форме ферритина – своеобразного резерва легкомобилизуемого железа. Кроме того, избыток железа может откладываться в тканях в виде хорошо известного морфологам метаболически инертного гемосидерина. Недостаток железа в организме может вызвать нарушение последнего этапа синтеза гема – превращение протопорфирина IX в гем. Как результат этого развивается анемия, сопровождающаяся увеличением содержания порфиринов, в частности протопорфирина IX, в эритроцитах. Микроэлементы. Большинство микроэлементов в крови находится в связанном с белками состоянии (медь - церрулоплазмин, цинк - карбоангидраза, йод–тироксин, селен - глутатионпероксидаза). Вопрос №148 Гемостаз и факторы свертывания крови. Тромбообразования и фибринолиз. Гемостаз - система, включающая в себя процессы:
Гемостаз проходит в 3 стадии:
Существует противосвертывающая система крови, которая направлена на ограничение распространения тромба местом повреждения сосуда. 1. Первичный гемостаз Только тромбоциты способны к адгезии и агрегации. Адгезия - налипание на края раны. Агрегация - скучивание вокруг раны. Тромбоциты должны быть активированы. Активация тромбоцитов заключается в:
В норме кровь не сворачивается, т.к. тромбоциты имеют пластинчатую форму, а не звездчатую, и не способны к агрегации. В кровеносных сосудах вырабатываются простациклины (производные арахидоновой кислоты), которые тормозят агрегацию тромбоцитов и сужение кровеносных сосудов. Для активации существуют первичные и вторичные индукторы активации:
Механизма активации тромбоцитов 1. При повреждении кровеносных сосудов тромбоцитами и эндотелием выделяется фактор фон Виллебранда (фВ), который взаимодействует с рецепторами тромбоцитов и коллагеном поврежденных сосудов, образует между ними мостики и способствует адгезии (прилипание к краям раны). Под действием фактора фон Виллебранда в тромбоцитах активируется фосфолипаза С (ФлС), которая стимулирует образование ИФ3, который стимулирует выведение Ca2+ из внутриклеточных депо. Ca2+ связывается с кальмодулином, и этот комплекс активирует миокиназу, которая путем фосфорилирования активирует сократительный белок тромбостенин. Он сокращается и изменяет форму тромбоцита с пластинчатой на звездчатую, что облегчает их сцепление между собой, т.е. агрегацию. Коллаген (появляется при повреждении кровеносных сосудов) взаимодействует с рецепторами тромбоцитов, активирует фосфолипазу А2, которая отщепляет от фосфолипидов мембран арахидоновую кислоту (20:4). Она под действием циклооксигеназы (ЦОГ) превращается в тромбоксаны, которые вызывают сужение сосуда и агрегацию тромбоцитов (агрегация пока обратимая, т.к при надавливании на края раны кровотечение восстанавливается) 2. Необратимая агрегация наступает под действием тромбина, который через ИФ3 высвобождает кальций из депо. Кальций активирует протеинкиназу С (ПкС), которая путем фосфорилирования активирует сократительный белок плекстрин. Он способен сокращать секреторные гранулы и высвобождать из них вторичные индукторы активации тромбоцитов. Под их действием происходит сужение сосудов и необратимая агрегация с образованием белого тромбоцитарного тромба. Кровотечение останавливается. В 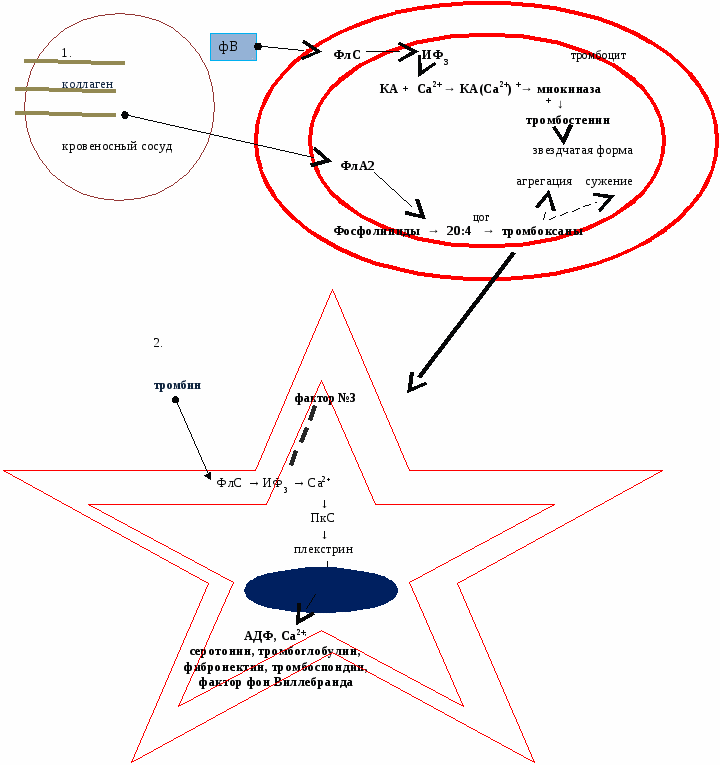 торичные индукторы активации тромбоцитов: торичные индукторы активации тромбоцитов:
Кроме того, при активации тромбоцитов на их поверхности появляются отрицательно заряженные фосфолипиды мембран - фактор №3. Эти участки тромбогеннные, т.к. на них будет протекать свертывание крови. Если диаметр кровеносного сосуда меньше 100 мкм, то свертывание крови заканчивается тромбоцитарным гемостазом. На ингибировании первичного гемостаза основано действие средств, "разжижающих" кровь (тромбоаз, аспирин - ингибирует ЦОГ → тормозится агрегация → снижается тромбообразование). Противоположное действие - коллагеновые гемостатические повязки, которые усиливают агрегацию, сужение кровеносных сосудов и, следовательно, более быстро останавливают кровотечение. Если повреждается более крупный сосуд, то наступает 2 стадия - гемокоагуляция. Происходит активация тромбокиназы, превращающей протромбин в тромбин. Это каскадный механизма, в результате которого проис ходит усиление сигнала. В нем принимают участие 13 факторов свертывания крови. Они находятся в неактивном виде, но при повреждении сосудов активируются частичным протеолизом, и к их номеру добавляется "а" - активированный. Ферментами являются II, VII, IX, X, XI, XII факторы. Все дальнейшие реакции с участием факторов свертывания крови протекают на мембранах тромбоцитов или клетках эндотелия поврежденных сосудов. М 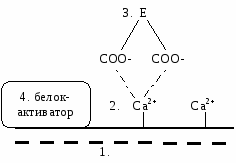 ембранные комплексы включают 4 компонента (на них происходит свертывание крови): ембранные комплексы включают 4 компонента (на них происходит свертывание крови):
Все ферменты имеют дополнительный отрицательный заряд (карбоксильную группу) в составе глутаминовой кислоты. Образуются γ-карбоксиглутаминовые кислоты (ГКГК) в печени с участием витамина К. Антивитамины К (дикумарол и варфарин) препятствуют карбоксилированию глутаминовой кислоты и, следовательно, свертываемости крови. Карбоксилирование глутаминовой кислоты В 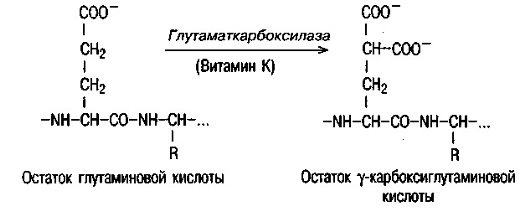 результате этого происходит активация мембранных комплексов. результате этого происходит активация мембранных комплексов.
2а - Прокоагулянтная стадия На первой стадии необходимо активировать тромбокиназу. Эта реакция происходит на мембранах тромбоцитов. Активация тромбокиназы Т 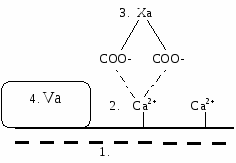 ромбокиназа - комплекс факторов: ромбокиназа - комплекс факторов:
Активация протекает двумя путями: 1 - прокоагулянтный (внешний) - 5-10 сек; инициатор - III фактор (тканевый); 2 - контактный (внутренний) - 10-12 мин; активируется при контакте XII фактора с коллагеном поврежденного сосуда. Менее распространен. Протекает возле воспаления на аномальных стенках (при атеросклерозе). 1- Внешний путь - каскадный (происходит усиление выработки тромбина). На мембранах поврежденных клеток эндотелия сосудов появляется первый мембранный инициирующий комплекс:
I 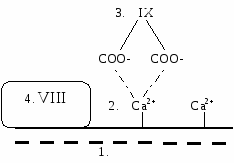 II фактор очень быстро активирует VII. II фактор очень быстро активирует VII.VIIа инициирует образование теназного мембранного комплекса. Теназный мембранный комплекс:
В  этом комплексе фактор IXа активирует тромбокиназу (фактор Х). этом комплексе фактор IXа активирует тромбокиназу (фактор Х).Х фактор катализирует превращение небольшого количества протромбина в тромбин. Тромбин по принципу обратной отрицательной связи вызывает активацию V, VII, VIII факторов в вышеперечисленных комплексах, что способствует каскадному усилению активации тромбокиназы. В результате под действием Х фактора образуется много тромбина. 2 - Внутренний путь. XII фактор при контакте с коллагеном активируется и образуется мембранный комплекс, который вместе с ВМК способен превращать прекалликреин в калликреин. Калликреин по принципу обратной отрицательной связи активирует XII фактор. В результате протромбин активируется частичным протеолизом и превращается в тромбин: 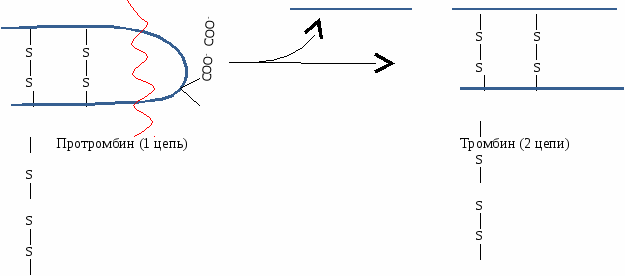 2б - Коагуляция Превращение фибриногена в фибрин под действием тромбина. Ф 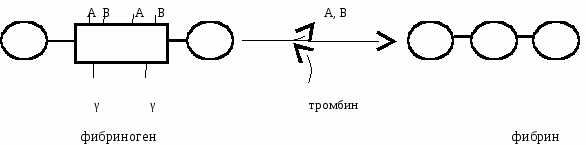 ибриноген состоит из 6 ппц (2А, 2В и 2γ). ибриноген состоит из 6 ппц (2А, 2В и 2γ).Отщепление отрицательно заряженных А и В концов способствует образованию фибрин-мономера, изменению его конформации, открытию участков взаимодействия с другими мономерами. В результате их агрегации образуется фибрин-полимер. Фибриновый сгусток рыхлый, в его структуре присутствует сыворотка и тромбоциты. Под действием XIII фактора происходит образование ковалентных связей между отдельными мономерами. 2в - Ретракция Под действием сократительного белка тромбостенина фибрин-полимер сжимается, из него выдавливается сыворотка. Образуется красный фибриновый тромб. который стягивает края раны, облегчая ее зарастание соединительной тканью. 3. Фибринолиз Разрушение красного фибринового тромба. Когда образовался тромб, в печени синтезируется плазминоген, который прикрепляется к тромбу вместе со своими активаторами. Активаторы плазминогена:
Под действием плазмина (активированного плазминогена) расщепляются фибриновые нити на мелкие кусочки (ппц), которые поступают в кровь. В результате тромб растворяется. Вопрос №149 Синтез гема и его регуляция. Г 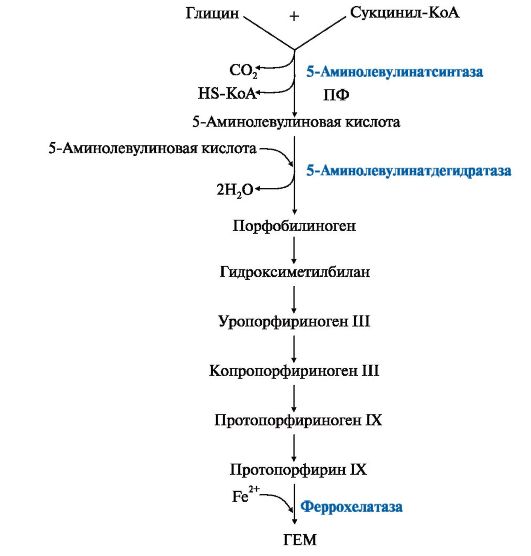 ем является простетической группой гемоглобина, миоглобина, цитохромов, каталазы, пероксидазы. Гем синтезируется во всех клетках, но наиболее активно синтез идет в печени и костном мозге. Эти ткани нуждаются в больших количествах гема, необходимого для образования гемоглобина и цитохромов. Субстратами синтеза гема являются глицин, сукцинил-КоА и Fe2+. В матриксе митохондрий из глицина и сукцинил-КоА под действием пиридоксальзависимого фермента 5-аминолевулинатсинтазы образуется 5-аминолевулиновая кислота, которая поступает в цитоплазму. В цитоплазме фермент 5 -аминолевулинатдегидратаза катализирует реакцию конденсации двух молекул 5-аминолевулиновой кислоты с образованием порфобилиногена. Далее из четырех молекул порфобилиногена последовательно образуются промежуточные метаболиты - порфириногены, последний из которых поступает в митохондрии и превращается в протопорфирин IХ. Фермент феррохелатаза завершает образование гема, присоединяя Fe2+ к протопорфирину IX. ем является простетической группой гемоглобина, миоглобина, цитохромов, каталазы, пероксидазы. Гем синтезируется во всех клетках, но наиболее активно синтез идет в печени и костном мозге. Эти ткани нуждаются в больших количествах гема, необходимого для образования гемоглобина и цитохромов. Субстратами синтеза гема являются глицин, сукцинил-КоА и Fe2+. В матриксе митохондрий из глицина и сукцинил-КоА под действием пиридоксальзависимого фермента 5-аминолевулинатсинтазы образуется 5-аминолевулиновая кислота, которая поступает в цитоплазму. В цитоплазме фермент 5 -аминолевулинатдегидратаза катализирует реакцию конденсации двух молекул 5-аминолевулиновой кислоты с образованием порфобилиногена. Далее из четырех молекул порфобилиногена последовательно образуются промежуточные метаболиты - порфириногены, последний из которых поступает в митохондрии и превращается в протопорфирин IХ. Фермент феррохелатаза завершает образование гема, присоединяя Fe2+ к протопорфирину IX.Д 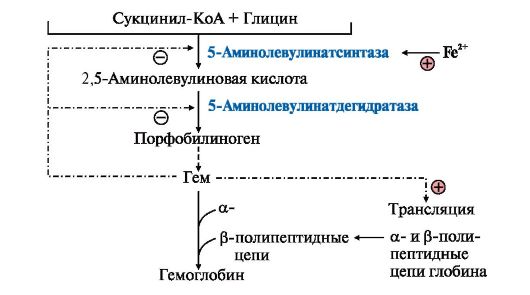 ве первые реакции синтеза гема катализируют ферменты, аллостерическим ингибитором которых является гем. Вместе с тем гем является индуктором синтеза α- и β-цепей гемоглобина. В ретикулоцитах Fe2+ индуцирует синтез 5-аминолевулинатсинтазы. Стероидные гормоны и некоторые лекарства (барбитураты, диклофенак, сульфаниламиды, эстрогены, прогестины) являются индукторами синтеза 5-аминолевулинатсинтазы. ве первые реакции синтеза гема катализируют ферменты, аллостерическим ингибитором которых является гем. Вместе с тем гем является индуктором синтеза α- и β-цепей гемоглобина. В ретикулоцитах Fe2+ индуцирует синтез 5-аминолевулинатсинтазы. Стероидные гормоны и некоторые лекарства (барбитураты, диклофенак, сульфаниламиды, эстрогены, прогестины) являются индукторами синтеза 5-аминолевулинатсинтазы. В результате генетических дефектов или нарушений регуляции ферментов, участвующих в биосинтезе гема, развиваются порфирии. Первичные порфирии обусловлены генетическими дефектами в структуре генов, кодирующих ферменты синтеза гема, вторичные - связаны с нарушениями регуляции реакций синтеза гема. Порфирии может вызвать прием лекарственных препаратов, являющихся индукторами синтеза 5-аминолевулинат- синтазы. Эти заболевания сопровождаются накоплением в клетках промежуточных метаболитов синтеза гема порфириногенов, которые оказывают токсическое действие на нервную систему и вызывают нейропсихические симптомы. Порфириногены на свету превращаются в порфирины, которые при взаимодействии с кислородом образуют активные радикалы, повреждающие клетки кожи. Анемия (малокровие) - уменьшение количества эритроцитов и (или) снижение содержания гемоглобина в единице объема крови. Анемия может быть как самостоятельным заболеванием, так и синдромом, сопровождающим течение другого патологического процесса. Распределение анемий на три группы: анемии вследствие кровопотери (постгеморрагические анемии); анемии вследствие нарушений процесса образования гемоглобина или процессов кроветворения; анемии, вызванные усиленным распадом эритроцитов в организме (гемолитические). Постгеморрагическая анемия возникает вследствие быстрой и массивной кровопотери. Причины - травмы (ранения) кровеносных сосудов с развитием внешнего кровотечения и внутренние кровотечения (желудочно-кишечные, кровотечения в брюшную полость, почечные, легочные, маточные, а также кровотечения из различных органов при геморрагических диатезах, внутренних заболеваниях). Механизм развития связан с резким сокращением общего объема крови в сосудах. Хроническая постгеморрагическая анемия развивается благодаря повторным незначительным кровопотерям, которые истощают запасы железа в организме. Железодефицитные анемии могут быть как внешними, так и внутренними. Основными факторами возникновения первых являются хронические кровопотери (вместе с эритроцитами теряется железо), повышенное расходование запасов железа (беременность, кормление, период роста детей). Вторые связаны с общим недостаточным питанием или длительным соблюдением диеты (особенно молочной) с ограниченным содержанием железа. Кроме того, отмечают нарушение всасывания железа при удалении желудка и кишечника, хроническом энтерите. При дефиците железа нарушается синтез гемоглобина, что приводит к задержке созревания клеток красной крови и выхода их в кровеносное русло. Сидероахрестические (железонасыщенные) анемии бывают наследственными и приобретенными. Наследственная форма связана с нарушением синтеза порфиринов, в частности протопорфирина, что ведет к снижению в эритроците количества гемоглобина и накоплению в организме несвязанного железа. Болеют чаще мужчины. В синтезе порфирина участвует витамин В6. В связи с этим выделяют В6-чувствительные и устойчивые формы этой анемии. В первом случае витамин В6 эффективен, во втором - нет. Приобретенные формы встречаются у лиц, имеющих контакт с рядом металлов (свинец, кадмий, никель), токсичных для человека. Длительный контакт приводит к связыванию групп ферментов аминокислоты, предшественницы протопорфирина и гемсинтетазы, в результате чего происходит накопление в эритроцитах железа, протопорфирина и его предшественников. Мегалобластная (витаминодефицитная анемия) при недостаточном поступлении в организм витаминов В12 и/или фолиевой кислоты. Дефицит этих витаминов приводит к нарушению в клетках синтеза ДНК и РНК, что вызывает нарушения созревания и насыщения гемоглобином эритроцитов. В костном мозге появляются крупные клетки - мегалобласты, а в периферической крови - крупные эритроциты (мегалоциты и макроциты). Процесс кроворазрушения преобладает над кроветворением. Неполноценные эритроциты менее устойчивы, чем нормальные, и гибнут быстрее. У взрослых людей распространена В12-дефицитная (пернициозная) анемия Аддисона-Бирмера, связанная с атрофией слизистой оболочки желудка. При этом происходит прекращение выработки слизистой оболочкой внутреннего фактора Кастла, который способствует всасыванию витамина В12, поступающего с пищей. Гемолитические - преобладание процессов кроворазрушения над процессами кровообразования. Кроворазрушение может происходить преимущественно внутри сосудов или вне их. Причины внутрисосудистого гемолиза: гемолитические яды, тяжелые ожоги, малярия, сепсис, переливание несовместимой крови, нарушения в работе иммунной системы, вирусные инфекции, хронический лимфолейкоз, системная красная волчанка. Внутриклеточный гемолиз происходит в некоторых внутренних органах, преимущественно в селезенке, и сопровождается увеличением селезенки. Вследствие повышенного разрушения эритроцитов в крови нарастает количество непрямого билирубина. У больных появляется желтушное окрашивание кожи и слизистых оболочек. При этом желчь и кал интенсивно окрашены вследствие значительного количества желчных пигментов билирубина и стеркобилиногена, моча темная за счет повышенного количества уробилина. Кроверазрушение сопровождается уменьшением общего количества эритроцитов и повышением количества ретикулоцитов в крови, а также увеличением количества эритробластов в костном мозге. Уровень железа сыворотки крови повышается. При ряде нарушений характерно снижение стойкости эритроцитов, что способствует их быстрому разрушению. При серповидноклеточной анемии отмечается замещение аминокислотной структуры цепей глобина с появлением HbS. В результате эритроциты приобретают форму серпа, веретена, иглы. При этом вязкость крови значительно увеличивается, скорость кровотока уменьшена, происходит закупорка мелких капилляров, сопровождающаяся тромбозами и инфарктами внутренних органов и тканей. При попадании серповидных эритроцитов в артериальную кровь форма их восстанавливается. Однако механическая устойчивость эритроцитов снижена, что приводит к их усиленному разрушению. Талассемия представляет собой целую группу гемоглобинопатии, определяемых различными генами. При этом заболевании гемоглобин А, свойственный взрослым, на 50-90% заменен на фетальный гемоглобин HbF. Он не способен продуктивно снабжать ткани кислородом, в результате чего развивается гипоксия, которая и приводит к резкому возрастанию производства эритроцитов. Усиление кровообразования может вызвать усиление всасывания железа, что приводит к сидерозу (отложению железа) органов. У аномальных эритроцитов сокращается продолжительность жизни, следовательно, увеличивается интенсивность распада крови. |
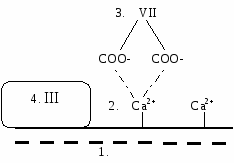 отрицательно заряженные фосфолипиды мембран;
отрицательно заряженные фосфолипиды мембран;