Микробиология. практикум микробиология ворд. Практикум министерство науки и высшего образования российской федерации
 Скачать 3.98 Mb. Скачать 3.98 Mb.
|

ОСНОВЫ ГЕННОЙ ИНЖЕНЕРИИ МИКРООРГАНИЗМОВСовременная микробиология тесно взаимодействует с биотех- нологией и генетикой. В частности, бактерии являются широко ис- пользуемыми платформами для наработки терапевтических бел- ков, а в свое время эксперименты на кишечной палочке помогли расшифровке генетического кода. Поэтому необходимо познако- мить студентов с рядом молекулярно-биологических и биотехноло- гических манипуляций с модельным организмом Escherichia coli(кишечной палочкой). Лабораторная работа № 11Выделение и очистка плазмидной ДНК методом щелочного лизисаОсновные теоретические положенияДля бактерий, кроме нуклеоида, характерно также наличие внехромосомных кольцевых ДНК – плазмид. Данные кольцевые молекулы имеют размеры от нескольких тысяч до нескольких де- сятков тысяч пар оснований. Они способны реплицироваться неза- висимо от деления бактериальной клетки и находятся в ней в коли- честве от нескольких до нескольких десятков штук. Кроме того, бактерии в ряде случаев способны обмениваться плазмидами друг с другом в процессах трансформации, конъюгации и трансдукции. Некоторые плазмиды, названные R-плазмидами (R – resistance), несут в себе гены устойчивости к антибиотикам. Обмен плазмида- ми между патогенными и непатогенными бактериями приводит к появлению и распространению множественной устойчивости к антибиотикам. Особенно это опасно в группе нозомикальных (внутрибольничных) инфекций. Однако способность плазмиды к независимой репликации, а также обмен плазмидами между клет- ками делают их удобным инструментом для генной инженерии. Плазмиды используют в молекулярной генетике в качестве векторов – молекул доставки необходимых исследователю генов в клетку. В дальнейшем это приводит к синтезу гетерологичных белков. Плазмида как молекула-челнок в молекулярной биологии обладает несколькими обязательными структурными элементами: Точка ori– точка начала репликации. Сайты рестрикции, служащие для разрезания плазмиды и ее модификации (подробнее об этом см. в лабораторной работе № 15). Ген устойчивости к антибиотику, позволяющий проводить селективное выращивание трансформированных клеток на среде с антибиотиком для проверки встраивания плазмиды. Маркерный ген (необязательная часть), служащий дополни- тельным индикатором успешной интеграции плазмиды в клетку. В качестве маркерных генов в последнее время все чаще выступают флуоресцирующие белки – в частности, зеленый флуоресцирую- щий белок (GFP). Его экспрессия позволяет визуально установить наличие плазмиды в клетке. Промотор и находящийся под его контролем целевой ген. До- ставка этих двух компонентов в клетку является основной целью всей процедуры. Выделение ДНК и манипуляции с ней – основа работ в моле- кулярной генетике. В зависимости от организма, из которого экстра- гируется ДНК, выделяют некоторые особенности реактивов и про- цедур. В любом случае в процессе выделения ДНК необходимо пройти несколько обязательных этапов: Лизис клеток – разрушение клеточной стенки, клеточной мембраны и других возможных оболочек клетки. Депротеинизация клеточного лизата – ферментативное рас- щепление белков или их осаждение. Центрифугирование в различных растворах для удаления из лизата мешающих компонентов на основе их растворимости. Осаждение ДНК. Очистка ДНК. Растворение ДНК в растворе для хранения, например, в TE-буфере. Помимо «ручных» методов, существуют методы выделения ДНК с использованием готовых растворов в специальных мини-колон- ках с сорбентом, селективно связывающим плазмидную ДНК. Та- кие коммерческие наборы чаще всего создаются для медицинских целей. Выделение ДНК возбудителя и последующая ПЦР с ней – один из самых быстрых способов обнаружить патоген. Выделение именно плазмидной ДНК основано на том, что при добавлении раствора 2 (см. ниже) pH поднимается до щелоч- ных значений (pH 12), и денатурации подвергается как неболь- шая плазмидная ДНК, так и крупная нуклеоидная (хромосомная) ДНК. Но крупная хромосомная ДНК денатурирует необратимо вследствие фрагментации ДНК в таких условиях, тогда как ДНК плазмиды при возвращении pH к нейтральным показателям при до- бавлении раствора 3 (см. ниже) ренатурирует. Довольно часто количественную и качественную оценку пре- парату выделенной ДНК в первом приближении дают при гель- электрофорезе, следующем, как правило, сразу за процедурой выде- ления. Для этого визуально сравнивают на соседних дорожках геля интенсивность свечения в ультрафиолете полученного образца с об- разцом известной концентрации. Определить концентрацию ДНК, а также степень ее чистоты можно с помощью спектрофотометра, что точнее и быстрее. Для этого измеряют оптическую плотность раствора ДНК при длине волны 260 нм. Одна (каждая) оптическая единица соответствует концентрации ДНК в 50 мкг/мл. Спектро- фотометрия (абсорбционная) – физико-химический метод иссле- дования растворов и твердых веществ, основанный на изучении спектров поглощения в ультрафиолетовой (200–400 нм), видимой (400–760 нм) и инфракрасной (> 760 нм) областях спектра. Основ- ная зависимость, изучаемая в спектрофотометрии, – зависимость интенсивности поглощения падающего света от длины. В сооответ- ствии с законом Бугера – Ламберта – Бера оптическая плотность раствора прямо пропорциональна концентрации поглощающего вещества. Нуклеиновые кислоты поглощают ультрафиолетовое (УФ) излучение в области 240–290 нм с максимумом при 260 нм. Хромо- форами служат азотистые основания нуклеиновой кислоты (НК), особенно пиримидиновые. Пиримидины поглощают УФ-свет при- мерно в 10–20 раз интенсивнее, чем хромофоры белковых молекул – триптофан, тирозин и фенилаланин. Для оценки чистоты препара- та ДНК, свободного от РНК, проводят измерения оптической плот- ности раствора при длинах волн 260, 280 и 235 нм, то есть на макси- мумах поглощения растворов ДНК, белков и полисахаридов соответ- ственно. Значение соотношения А260/280 для чистой ДНК должно быть больше, чем 1,8, значение А260/235 – больше, чем 2,2. Цель работы: научиться выделять внехромосомную ДНК бактерий для последующих рестрикции, электрофореза и транс- формации; изучить количество выделенной плазмидной ДНК и ее чистоту. Оборудование и реактивыМикроцентрифуга. Культура бактерий с плазмидой. рН-метр. Аналитические весы. Вортекс. Реактивы: глюкоза, ЭДТА, додецилсульфат натрия (ДСН, SDS), гидроксид натрия, трис-аминометан, ацетат калия, соляная кисло- та, лизоцим, изопропанол. Пластиковые микропробирки. Автоматические пипетки и наконечники для них. Микропланшетный спектрофотометр TecanМ200Pro. Микропланшет NanoQuant. Марлевые салфетки и вата. Этанол. Дистиллированная вода. Буфер ТЕ. Ход работыПриготовить растворы для лизиса: Раствор 1 (10 мл): 10 мМ ТрисHCl, pH 7,4, 5 мМ ЭДТА, 50 мМ глюкозы, 2 мг/мл лизоцима. Раствор 2 (10 мл): 0,2 М NaOH, 1 % SDS. Раствор 3 (10 мл): 3 М ацетат калия. Налить в пробирки типа эппендорф по 100 мкл раствора 1. Подписатьэппендорфы! В стерильных условиях собрать стерильной петлей с чашки Петри небольшое количество бактериальной культуры. Перенести в эппендорф с раствором 1. ИЛИ Осадить клетки из культуральной среды. Для этого поместить 1,5 мл «ночной культуры» E. coli в 1,7 мл микроцентрифужную пробирку и центрифугировать в течение 2 мин при 10 000 об/мин. Удалить супернатант выливанием через край. Повторить осаждение в эту пробирку еще два раза. Подписатьэппендорфы! Центрифугировать 10 с дополнительно, отобрать остатки куль- туральной среды микропипеткой. Добавить 100 мкл раствора 1. Ресуспендировать культуру в буфере до образования гомо- генной суспензии. Закрыть крышку, 2–3 мин перемешивать гомо- генат, переворачивая пробирку в руках. При этом происходит раз- рушение клеточной стенки под действием лизоцима. Вортекс ис- пользовать не рекомендуется. Добавить 200 мкл (двойной объем) раствора 2. Закрыть крыш- ку эппендорфа и аккуратно перемешать легким встряхиванием или перевернуть эппендорф несколько раз. Вортекс использовать не ре- комендуется, так как плазмида может порваться. Выдержать 10 мин. при комнатной температуре. Происходят лизис бактерий и щелочная денатурация ДНК. Добавить 150 мкл (1,5 объема) охлажденного раствора 3. Ак- куратно перемешать встряхиванием. Оставить на 10 мин в холо- дильнике при +4 – +8 °С. Поместить эппендорфы в микроцентрифугу, осадить в тече- ние 5 мин. при максимальных оборотах (14 500 об./мин.). Супернатант, содержащий плазмидную ДНК, аккуратно, не за- девая осадок, отобрать и перенести в чистую подписанную пробирку. Добавить 1 мл холодного изопропанола. Аккуратно пере- мешать. Осаждение проводить в течение 1 ч при –20 °С. Осадить плазмиду центрифугированием в течение 10 мин. при максимальной скорости центрифуги. Супернатант вылить че- рез край, его остатки после короткого центрифугирования отобрать микропипеткой. Добавить 300 мкл 70 % этилового спирта для отмывания осадка от соли. Перемешать и центрифугировать 2 мин. Удалить спирт. Осадок подсушить на воздухе. Нужно избегать пересушива- ния осадка, так как это снижает его растворимость. Растворить осадок ДНК в 25–50 мкл ТЕ-буфера. Проверить количество и чистоту выделенной ДНК с помощью электрофореза в агарозном геле (10 мкл, лабораторная работа № 12) или на спектрофотометре. Внести в рабочую тетрадь основные принципы выделения ДНК хромосомной и плазмидной. Спектрофотомерия препаратов ДНКВключить персональный компьютер и спектрофотометр. Кнопка включения прибора находится на задней стенке. Когда при- бор включен, на его верхней панели в правом нижнем углу горит зеленый индикатор. Запустить программу I-control 1–10. Когда программа загру- зится, зайти на панели управления на вкладку Instrument, нажать кнопку Connectи выбрать в появившемся окне установленную мо- дель прибора. Нажать ОК. Теперь прибор готов к работе. В нижнем левом углу открывшегося диалогового окна вы- брать закладку Applications. Автоматически выбрана программа для определения качества и количества нуклеиновых кислот с ис- пользованием планшета NanoQuant. В выпадающем меню Tipe выбрать тип образца, который нужно проанализировать: деспирализированная геномная ДНК (dsDNA), РНК (RNA), суперспирализированная плазмидная ДНК (ssDNA). Для работы необходим вариант ssDNA. Провести калибровку прибора. Для этого взять планшет, снять крышку. Нанести на каждую из лунок по 2 мкл буфера ТЕ и накрыть планшет крышкой. Поставить планшет в прибор, нажав на зеленую кнопку на верхней поверхности спектрофотометра. В диалоговом окне нажать кнопку Start Blanking. После заверше- ния калибровки в верхней части диалогового окна кнопка Startначнет подсвечиваться зеленым цветом. Подготовить планшет к измерению. Для этого открыть его, стереть сухим ватным тампоном калибровочный буфер, удалить остатки буфера, протерев планшет изнутри тампоном, смоченным в воде, а после – тампоном, смоченным в этаноле. Дать испарить- ся этанолу. Нанести образцы для анализа. Вносить по 2 мкл в лунку, ис- пользуя новый наконечник для каждого образца. Поместить план- шет в прибор, нажать в диалоговом окне кнопку Start. Через не- сколько минут измерение будет закончено, после чего автоматичес- ки откроется электронная таблица, в которой будут указаны номер лунки, концентрация нуклеиновых кислот в данной лунке (в нг/мкл, что соответствует мкг/мл), соотношение оптических плотностей при длине волны 260 нм и 280 нм. Это показатель качества и чис- тоты выделение ДНК. Он должен быть больше, чем 1,8. Очистить планшет от образцов ДНК с помощью ватного там- пона, воды и этанола, как указано в п. 6, и убрать его в футляр. Сохранить результаты на флеш-карте, закрыть программу, отключить прибор и компьютер. Вопросы для самоподготовкиКакие основные участки входят в плазмиду, какие функции они выполняют? Какие стадии выделения ДНК вы знаете? В чем принцип выделения плазмидной ДНК? Какую роль выполняют плазмиды в бактериальной клетке? Как их можно использовать в биотехнологии? Для чего используют фенол и хлороформ при выделении ДНК? Что такое лизоцим? Для чего его добавляют в раствор 1? Лабораторная работа № 12 Электрофорез плазмидной ДНК в агарозном геле Основные теоретические положенияМетод электрофореза широко используют в биологии и меди- цине для разделения заряженных молекул биополимеров – белков и нуклеиновых кислот. Суть метода заключается в том, что заряжен- ные частицы в растворе под действием электрического поля начи- нают двигаться. При помещении на их пути трехмерных сетчатых структур – гелей (акриламидного, агарозного, крахмального) ско- рость движения частиц замедляется пропорционально их размеру (массе), что и приводит к их разделению. Более маленькие (а зна- чит, и легкие) молекулы движутся быстрее, чем молекулы больше- го размера. ДНК – слабая кислота и под действием электрического тока двигается к аноду, заряженному положительно. На первом этапе электрофореза необходимо приготовить ага- розный гель. Готовится он добавлением агарозы в TAE буфер. Для полного растворения агарозы смесь нагревают до 95 °С. Пока гель находится в жидком состоянии, в него добавляют бромистый этидий и разливают в форму, не забывая ставить гребенку для обра- зования лунок. Бромистый этидий впоследствии встроится (ин- теркалирует) между парами нуклеотидов, что позволит визуализи- ровать ДНК в ультрафиолетовом свете. Очень важна на этом эта- пе концентрация агарозы. Она может варьировать от 0,3 до 2,0 %. Чем больше концентрация агарозы, тем меньше образующиеся по- ры и тем меньшего размера молекулы ДНК гель способен разделять. Следующим этапом электрофоретического разделения являет- ся внесение образцов ДНК в лунки. Для этого используют буфер при внесении образцов в гель. Он содержит глицерин или сахарозу, необходимые для того, чтобы ДНК сразу опустилась на дно лунки, и краситель, позволяющий отследить движение электрофоретичес- кого фронта. Параллельно с образцами обычно в одну из лунок вно- сится маркер ДНК – смесь нуклеотидов известной длины (рис. 18). Маркер служит показателем успешности электрофореза. 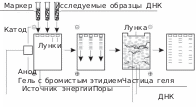 Длинные фрагменты Длинные фрагментыКороткие фрагменты Рис. 18. Процесс электрофореза После внесения всех образцов в гель включают источник тока, и начинается непосредственно электрофорез. В процессе электро- фореза более короткие фрагменты проще проходят через порис- тую структуру геля и оказываются ближе к аноду (рис. 18). Конечным этапом работы является визуализация результатов в трансиллюминаторе в УФ-спектре. Цель работы: освоить метод электрофореза нуклеиновых кислот в агарозном геле. Установить качество выделения плаз- мидной ДНК. Оборудование и реактивыБуфер для электрофореза – ТАЕ ( 50). Агароза. Бромистый этидий (Внимание!Данное вещество является канцерогеном! Работать только в перчатках! Не проливать на стол!). Буфер для внесения с красителем бромфеноловым синим. Камера для горизонтального электрофореза с заливочным столиком. Источник постоянного тока. Электроплитка. Автоматическая пипетка с наконечниками. Трансиллюминатор. Ход работыПриготовить буфер для электрофореза из стока (концентра- та). Для этого взять 20 мл ТАЕ ( 50) и довести дистиллированной водой до 1 л. Приготовить агарозный гель. Для этого взять 1 г агарозы, по- местить в коническую колбу объемом 250 мл, добавить 100 мл бу- фера ТАЕ ( 1). Довести буфер до кипения, дождаться полного раст- ворения агарозы. После ее охлаждения до 70 °C добавить 5 мкл раствора бромистого этидия (1 % раствор) и тщательно перемешать. Собрать заливочный столик, выровнять его, поставить гре- бенку. Залить расплавленную агарозу ровным слоем, избегая по- явления пузырьков воздуха. Дождаться полимеризации и остывания геля, аккуратно вы- нуть гребенку. Достать застывший гель из формы, поместить его в электрофоретическую камеру, предварительно заполненную 1 TAE-буфером. К 5–10 мкл выделенной плазмидной ДНК добавить 0,5–1 мкл буфера, перемешать, аккуратно внести в лунки геля. После внесения образцов закрыть камеру крышкой, подклю- чить электроды к источнику тока. Включить электрический ток, выставив на источнике напря- жение от 60 до 90 В и силу тока 20–40 мА. Проводить электрофорез в течение 1–1,5 ч. Отключить напряжение, достать гель (вперчатках!) и пере- нести на стекло трансиллюминатора. Закрыть защитной крышкой и включить ультрафиолет. Для начала убедится в успешности элек- трофоретического разделения, просмотрев разделение маркера. Пронаблюдать светящиеся полосы: рядом с лунками – плазмид- ная ДНК, облака на противоположной стороне – продукты дегра- дации РНК. Внести в рабочую тетрадь основные принципы проведения электрофоретического разделения ДНК в агарозном геле. Вопросы для самоподготовкиКакие типы гелей используются в электрофорезе биополимеров? Какие они имеют свойства, достоинства и недостатки? Как возможно визуализировать ДНК в геле, после электрофореза? Для чего используют буфер для внесения и маркер молекулярных масс? Почему для электрофореза и приготовления геля используют специальные буферы, а не дистиллированную воду? Почему буфер имеет щелочную рН, а не кислую? Каким методом, кроме электрофореза, можно установить качество выделенной ДНК и ее концентрацию? |