современные химические методы анализа. Тема 1 Методы аналитической химии
 Скачать 465.58 Kb. Скачать 465.58 Kb.
|

|
Иммунный анализ Иммунитет – способ защиты организма от генетически чужеродных веществ как живой, так и неживой природы, направленный на сохранение структурной и функциональной целостности организма и его биологической индивидуальности (качественного гомеостаза). Антигены – структурно (генетически) чужеродные для конкретного организма вещества, способные вызвать иммунный ответ, направленный на их удаление. Антигены могут быть экзогенными (попадать в организм из окружающей среды) и появляться в собственном организме (эндогенные), например, антигены опухолевых клеток, действия радиоактивного излучения. Иммунная система – совокупность органов, тканей и клеток, которая обеспечивает поддержание антигенного гомеостаза (постоянства) организма путем распознавания и элиминирования чужеродных антигенов. Защита организма от генетически чужеродных веществ живой и неживой природы – антигенов, т. е. поддержание гомеостаза осуществляется двумя группами факторов: – факторы, обеспечивающие неспецифическую устойчивость (резистентность) организма к антигенам независимо от их происхождения; – специфические факторы иммунитета, которые направлены против конкретных антигенов. Антитела – белки, способные к специфическому связыванию с антигенными детерминантами (эпитопами) антигенов. Антитела относятся к белкам семейства g-глобулинов (иммуноглобулинов). Иммунный ответ – реакция иммунной системы организма, индуцированная антигеном и направленная на его разрушение и удаление из организма. Иммунный ответ зависит от свойств антигена, иммунологической реактивности организма и условий среды. В основе иммунохимических методов анализа лежит иммунная реакция взаимодействия антигена с антителом. При взаимодействии специфических антител с антигеном между аминокислотными остатками антигенсвязывающего центра (паратопа) антитела и антигенной детерминантой (эпитопом) антигена возникают взаимодействия: водородные, электростатические, Ван-дерВаальсовы, гидрофобные. Афинность (сродство) – степень специфического сродства антигенсвязывающего центра антитела (паратопа) к антигенной детерминанте (эпитопу) антигена – это результирующая сила возникающих между антигеном и антителом сил притяжения и отталкивания. Соединение антигена с антителом обратимо и к реакции взаимодействия антиген – антитело можно применить закон действующих масс и определить константу равновесия (Кр), которая и представляет собой константу аффинности (сродства). Аффинность относится к связыванию антитела с одной антигенной детерминантой (эпитопом) или моновалентным гаптеном. Поливалентность антигена и антител усиливает прочность соединения, так как для диссоциации комплексов антиген – антитело необходим разрыв всех связей. Авидность – суммарная сила взаимодействия антитела с антигеном. Чем выше аффинность (степень сродства), тем выше и авидность. Авидность тем сильнее, чем больше связывающих центров. 13 Реакции преципитации (от лат. precipitation – стремительное падение вниз) основаны на формировании и осаждении (выпадении в осадок) комплекса – растворимый молекулярный антиген – антитело – в присутствии электролита. Различают несколько разновидностей реакций преципитации: реакции кольцепреципитации; радиальная иммунодиффузия по Манчини (метод, позволяющий количественно определять антигены и антитела); иммуноэлектрофорез (сочетание метода электрофореза и иммунопреципитации). Эти методы носят чаще качественный или полуколичественный характер. При очень низких концентрациях компонентов образование комплекса антиген – антитело нельзя зафиксировать визуально и с помощью простых инструментальных средств. Тогда индикацию комплекса антиген – антитело проводят, вводя в один из компонентов – антиген или антитело – метку: радиоактивные изотопы (например, 125I), флюоресцентные вещества, ферменты. В зависимости от метки различают радиоиммуный (РИА), флюоресцентный иммунный (ФИА), иммуноферментый (ИФА) методы анализа. В последние годы широкое практическое применение получил ИФА, что связано с возможностью количественных определений, высокой чувствительностью, специфичностью и автоматизацией анализа. ИФА – группа методов, которые позволяют определить комплекс антиген – антитело с помощью субстрата, который расщепляется ферментом с появлением окраски. Суть метода состоит в соединении компонентов реакции антиген – антитело с измеряемой ферментной меткой. Антиген или антитело, вступающие в реакцию, метятся ферментом. По превращению субстрата под действием фермента можно судить о количестве вступившего во взаимодействие компонента реакции антиген – антитело. Фермент служит маркером иммунной реакции, позволяет наблюдать ее визуально или инструментально. Ферменты – удобные метки, их каталитические свойства позволяют им действовать в качестве усилителей, так как одна молекула фермента может способствовать образованию более 1´105 молекул продукта каталитической реакции в минуту. Основные способы получения антител или антигенов, меченых ферментом, – конъюгатов: химические, иммунологические и генно-инженерные. Наиболее часто применяют ферменты пероксидаза хрена, щелочная фосфатаза, галактозидаза. Для выявления активности фермента в комплексе антиген-антитело используют хромогенные субстраты, растворы которых изначально бесцветные, в процессе ферментативной реакции приобретают окраску, интенсивность которой пропорциональна количеству фермента. Так, для выявления активности пероксидазы хрена в твердофазном ИФА в качестве субстрата используют 5-аминосалициловую кислоту, дающую интенсивное коричневое окрашивание, орто-фенилендиамин, образующий оранжево-желтое окрашивание; для выявления активности щелочной фосфатазы и βгалатозидазы используют нитрофенилфосфаты и нитрофенилгалактозиды соответственно. Результат реакции при образовании окрашенного продукта 14 определяют визуально или с помощью спектрофотометра, измеряющего поглощение света с определенной длиной волны. Известно несколько вариантов постановки ИФА. Различают гомогенный и гетерогенный варианты. По методике постановки разделяют конкурентный и неконкурентный методы ИФА. Если на первой стадии в системе присутствуют только анализируемое соединение и соответствующие ему центры связывания (антиген и специфические антитела), то метод является неконкурентным. Если на первой стадии присутствуют анализируемое соединение (антиген) и его аналог (меченый ферментом антиген), конкурирующие между собой за связывание с имеющимися в недостатке центрами специфического связывания (антителами), то метод является конкурентным. В этом случае чем больше исследуемого антигена содержит раствор, тем меньше количество связывающихся меченых антигенов. Перспективные направления развития иммуноферментного анализа – создание экспресс-методов, основанных на использованием мембранных и иммунохроматографических систем, проточно-инжекционных методов, кинетического анализа, иммунобиосенсорных устройств, позволяющих проводить экспресс-анализ, в том числе одновременное определение нескольких антигенов в одном образце в реальном времени. Некаталитические реакции в кинетических методах анализа Некаталитические методы позволяют проводить однокомпонентный анализ и определять индивидуальные компоненты в смеси. Для определения как органических, так и неорганических веществ при сравнительно больших концентрациях можно использовать некаталитические реакции, протекающие с измеримой скоростью. Это могут быть реакции окислительновосстановительные, гидролиза, замещения лигандов, образования осадков. Их скорость или индукционный период определяется концентрацией реагирующих веществ в растворе. Определить тот или иной компонент в смеси можно, если скорость его взаимодействия с аналитическим реагентом существенно отличается от скорости реакции других веществ. Если два сходных по химическим свойствам соединения А и В реагируют с одним и тем же реагентом R, то возможно определить в начальный период реакции вещество А на фоне В с погрешностью < 1%, если k1/k2 > 500 k1 и k2 – константы скорости реакций. Можно проводить анализ смесей органических соединений (спиртов, сахаров, аминов) и смесей ионов металлов (щелочноземельные и редкоземельные элементы). Некаталитические методы не являются высокочувствительными: сmin составляет 10-2 – 10-1 мкг/мл или 10-6 – 10-4 М. Чувствительность определяется методом наблюдения за скоростью индикаторного процесса. Методы селективны. Методы и приемы измерения скоростей некаталитических реакций те же, что и каталитических. Если имеется подходящий равновесный или статический метод, то для анализа однокомпонентной системы не рекомендуют применять кинетический (динамический) метод, поскольку точность такого кинетического метода ниже, чем равновесного (статического). В целом количественные определения кинетическим методом с использованием некаталитических реакций используют редко. Но в ряде случаев они предпочтительны, например, при анализе органических смесей. Преимущество кинетического некаталитического метода в его простоте, возможности обходиться без сложной аппаратуры. Каталитические и ферментативные методы анализа Первый случай кинетического определения вещества с использованием каталитического эффекта описан Гейярдом в 1876 году – определение ванадия по его каталитическому действию в реакции окисления анилина хлоратом калия. Реакции, катализируемые веществами, которые надо определить, называют индикаторными. Чтобы индикаторная реакция была пригодна для анализа, ее скорость должна быть очень низкой или пренебрежимо малой по сравнению со скоростью катализируемой реакции. Широкое распространение кинетических методов в последние годы обусловлено улучшением их чувствительности (сmin лежит в интервале 10-6 – 10- 5 мкг/мл или 10-12 – 10-10 М и ограничена колебаниями «фона» матрицы) и селективности именно за счет использования каталитических реакций. Чувствительность каталитических методов (для неорганических веществ) сравнима с чувствительностью масс-спектральных и активационных методов анализа, для органических — с высокоэффективной жидкостной хроматографией. При определении серебра, хрома, кобальта, ртути каталитические методы наиболее чувствительны из всех известных методов анализа. Преимущество каталитических методов - простота аппаратурного оформления и методики проведения анализа. Среди каталитических методов высокую чувствительность и селективность имеют ферментативные методы. Ферментативными методами определяют субстраты, сами ферменты и эффекторы ферментов. Методы определения субстратов (веществ, на которые действуют ферменты) 9 высокоселективны (специфичны), позволяют провести определение субстратов непосредственно в матрице сложных объектов (кровь, биомассы и биожидкости, многокомпонентные технологические растворы). Чувствительность определения обусловлена методом, выбранным для контроля за скоростью процессов. Часто используют ферментные электроды. Методы определения эффекторов ферментов высокочувствительны, но не всегда селективны. В кинетических методах наиболее часто используют метод тангенсов как наиболее точный (использует большое число экспериментальных данных) и универсальный (применим для реакций с индукционным периодом). Реже применяют способ фиксированного времени и способ фиксированной концентрации, хотя эти способы более просты и менее трудоемки. Способ фиксированной концентрации используют обычно при автоматизации контроля, способ фиксированного времени — при проведении серийных анализов. Каталитические методы позволяют проводить определение самого катализатора, а также эффекторов катализа (активаторов и ингибиторов). Эффекты, изменяющие скорость реакций, имеют теоретическое и практическое значение и ассоциируются с каталитическими реакциями (главной областью применения в аналитической химии), хотя могут быть использованы также в некаталитических и фотохимических реакциях. Скорость любой каталитической реакции можно увеличить или уменьшить («активировать» или «ингибировать» реакцию) путем введения подходящих веществ. Это позволяет решать практические задачи, требующие крайне низкие пределы обнаружения (менее 10-12 М). Они достижимы лишь при использовании явления активирования. Важно, что появляется возможность определения активатора, который сам по себе чаще всего не обладает каталитическими свойствами. Явление ингибирования позволяет определять некоторые вещества, выступающие в качестве ингибиторов каталитических реакций. Модификатор можно определить при условии, что скорость модифицированной реакции пропорциональна его концентрации. Часто ингибиторы определяют с помощью каталитического титрования. В ферментативных реакциях активирование (ингибирование) является следствием присутствия ионов металлов, которые можно определить по их влиянию на каталитические свойства фермента. Скорость неферментативных реакций модифицируют с помощью гидроксилсодержащих, полиаминокарбоксильных и хромогенных лигандов. Комплексоны чаще используют как ингибиторы, другие лиганды могут быть и ингибиторами, и активаторами. Неорганические анионы часто являются ингибиторами катализируемых металлами реакций, в то время как ионы металлов чаще используют для активирования реакций, катализируемых металлами. Большинство металлов являются катализаторами в тех индикаторных реакциях, которые используют для их определения. Для того, чтобы расширить круг определяемых компонентов, каталитические процессы применяют также для определения активаторов и особенно ингибиторов, то есть соединений, соответственно увеличивающих или уменьшающих действие катализатора. Так, например, для определения щелочноземельных металлов каталитическим методом применяют принцип конкурентного комплексообразования. При этом в индикаторную реакцию, катализируемую медью (II), вводят ингибитор этилендиаминтетраацетат (ЭДТА) до практического прекращения протекания реакции. Если в такую систему затем добавить ион одного из щелочноземельных элементов (Ca, Sr, Ba) или ионы, не проявляющие, как правило, собственного каталитического действия (Cd, Zn), то по мере добавления ионов таких металлов они, образуя комплексные соединения с ЭДТА, высвобождают ионы меди, и скорость каталитического индикаторного процесса увеличивается. При этом увеличение скорости пропорционально концентрации ионов металлов, добавленных в катализируемую медью (II) реакцию. Активирующее и особенно ингибирующее действие используют для определения органических соединений самых разных классов. Органические соединения и сами могут быть катализаторами в индикаторных системах (альдегиды, N-нитрозоамины, серосодержащие органические соединения), но чаще их определяют по активирующему или ингибирующему действию. Например, катализируемую медью реакцию окисления гидрохинона пероксидом водорода применяют для определения 10-10-10-9 моль/л карбоксил-, азот- и фосфорсодержащих комплексонов самого разного строения. Часто определяют каталитическими методами очень малые количества селена, теллура и таких анионов, как I- , F, CN- , S2-. Лекция № 3. Электрохимические методы анализа Электрохимия – раздел физической химии, который рассматривает системы, содержащие ионы (растворы или расплавы электролитов) и процессы, протекающие на границе двух фаз с участием заряженных частиц. Первые представления о взаимосвязи химических и электрических явлений были известны в XVIII веке, так как было выполнено огромное количество физико-химических экспериментов с электрическим и грозовыми разрядами, с зарядами, находящимися в лейденских банках, но все они имели случайный характер из-за отсутствия постоянного мощного источника электрической энергии. Зарождение электрохимии связано с именами Л. Гальвани и А. Вольта. Занимаясь исследованием физиологических функций лягушки, Гальвани случайно создал электрохимическую цепь. Она состояла из двух различных металлов и препарированной лапки лягушки. Лапка одновременно являлась электролитом и индикатором электрического тока, но вывод был дан неправильный, т. е., согласно Гальвани, этот электрический ток, который возникал в цепи, имел животное происхождение, т. е. был связан с функциональными особенностями организма лягушки (теория «животного электричества»). Правильное толкование опытам Гальвани дал А. Вольта. Он создал первую батарею гальванических элементов – вольтов столб. Элементы батареи состояли из медных и цинковых дисков, а электролитом служил пропитанный соленой водой или кислотой губчатый материал. Именно такое соединение позволило получить электрический ток. Вскоре трудами великих ученых А. Вольта, Дж. Даниэля, Б. С. Якоби, П. Р. Багратиона, Г. Плантэ и др. появились удобные в работе мощные гальванические элементы и аккумуляторы. Затем А. Вольта разработал ряд напряжений металлов. Если два различных металла привести в соприкосновение, а затем разъединить, то при помощи физических средств, например, электроскопа, можно увидеть, что один металл приобрел положительный заряд, а другой – отрицательный. Этот ряд металлов, в котором каждый предшествующий металл заряжается положительно, но после контакта с любым последующим, т. е. ряд Вольта, оказался аналогичным ряду напряжений. Далее, в начале XIX века, был разработан электролиз, а М. Фарадей установил количественные законы электролиза. Большой вклад в развитие электрохимии внесли ученые: С. А. Аррениус, В. Ф. Оствальд, Р. А. Колли, П. Дебай, В. Нернст, Г. Гельмгольц и др. Сейчас электрохимия делится на теоретическую и прикладную. Благодаря использованию электрохимических методов, она связана с другими разделами физической химии, а также с аналитической химией и другими науками. потенциометрия кондуктометрия кулонометрия ЭЛЕТРОХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ Необходимость в использовании разнообразных методов для исследования электрохимических процессов обусловлена широкой областью изменения скорости переноса электрона в электродных реакциях. Каждый из методов имеет некоторый предел по определяемому значению плотности тока обмена, выше которого электрохимические параметры электродной реакции определить нельзя. Применительно к каждому конкретному объекту необходимо выбрать тот метод, который дает максимальный объем надежной информации. При проведении электрохимических исследований необходимо знать химический состав исходных веществ и продуктов реакции. Для определения состава электролита используют различные физико-химические методы: спектрофотометрический, потенциометрический, аналитический и другие. При проведении электрохимических исследований необходимо соблюдать следующие условия. 1. Максимальная чистота используемых реактивов; состав электродов должен быть строго известен, как известно и состояние их поверхностей. Следует следить за тем, чтобы в процессе измерений поверхность электродов не претерпевала изменений. 2. Конструкция электрохимической ячейки и расположенный в ней электродов должны обеспечивать равномерное распределение тока по всей поверхности рабочего электрода. 3. Измерение проводить при строго контролируемой температуре. 4. Поддерживать постоянные давления и состав газовой фазы над электролитом. Как правило, исследования проводят в среде инертного газа (N2, Ar, Ne, He H2), поскольку кислород газовой фазы может оказывать существенное влияние на механизм процесса. 5. Необходимо обеспечить такие условия эксперимента, при которых падение потенциала в диффузной части двойного электрического слоя было бы минимальным или точно известным. Для снижения этого потенциала используют, как правило, фоновый электролит, концентрация которого должна быть не менее, чем в 20 раз выше, чем у основного вещества. Однако предварительно следует убедиться, что фоновый электролит не искажает поляризационной кривой изучаемой реакции. 6. Точное измерение потенциала рабочего электрода. Для этого необходимо устранить диффузионный потенциал между исследуемым электролитом и электролитом электрода сравнения. Этот потенциал принимает максимальное значение при приближении к предельному току и может, существенно исказить результаты измерений. Для устранения диффузионного потенциала между исследуемым электролитом и электролитом электрода сравнения желательно: а) выбрать электрод сравнения, который имеет тот же электролит по составу, что и исследуемый. Например, при исследованиях в хлоридных растворах удобно применять хлор-серебрянный, каломельный, хлорный электроды; в кислых сульфатных растворах – ртутно-сульфатные электроды и т.п.; б) использовать электрод сравнения с таким электролитом, на границе которого с исследуемым электролитом диффузионный потенциал может быть рассчитан по известным формулам. При измерении в растворах с постоянной ионной силой, а при больших концентрациях фона – с постоянной ионной концентрацией можно, в принципе, использовать любой электрод сравнения. Диффузионный потенциал в этом случае может быть и весьма велик, но и постоянен – его можно рассчитать или определить экспериментально. Во всех случаях изучения кинетики электрохимических процессов необходимо измерение плотности тока. Обычно начинают с того, что выясняют методами аналитической химии, кулонометрии, протекает ли на электроде только одна изучаемая реакция или она осложнена побочными. В случае протекания побочных реакций, надо выяснить, какая доля тока приходится только на осуществление изучаемой реакции (построить так называемую парциальную поляризационную характеристику для изучаемой реакции). Наиболее просто механизм электродной реакции можно интерпретировать лишь в случае, когда исходное вещество превращается в один продукт со 100%-ным выходом по току. Проверка реакции на соответствие закону Фарадея или проведение кулонометрических измерений позволяет одновременно определить число электронов, участвующих в суммарной электродной реакции. Знание состава исходного вещества и продукта реакции, а также общего числа переносимых электронов, дает возможность записать уравнение суммарной электродной реакции. Следующим шагом в изучении механизма электродной реакции является выяснение того, какая стадия является лимитирующей. Если лимитирующей стадией является стадия разряда -ионизации, а все другие протекают обратимо, то основные кинетические параметры процесса можно определить графически или аналитически, применяя к поляризационным характеристикам уравнения теории замедленного разряда [1]. Электрохимические методы анализа Электрохимические методы анализа – это совокупность методов качественного и количественного анализа, основанных на электрохимических явлениях, происходящих в исследуемой среде или на границе раздела фаз и связанных с изменением структуры, химического состава или концентрации анализируемого вещества. Разновидностями метода являются электрогравиметрический анализ (электроанализ), внутренний электролиз, контактный обмен металлов (цементация), полярографический анализ, кулонометрия и др. В частности, электрогравиметрический анализ основан на взвешивании вещества, выделяющемся на одном из электродов. Метод позволяет не только проводить количественные определения меди, никеля, свинца и др., но и разделять смеси веществ. Кроме того, к электрохимическим методам анализа относят методы, основанные на измерении электропроводности (кондуктометрия) или потенциала электрода (потенциометрия). Некоторые электрохимические методы применяются для нахождения конечной точки титрования (амперометрическое титрование, кондуктометрическое титрование, потенциометрическое титрование, кулонометрическое титрование). Различают прямые и косвенные электрохимические методы. В прямых методах используют зависимость силы тока (потенциала и т.д.) от концентрации определяемого компонента. В косвенных методах силу тока (потенциал и т. д.) измеряют с целью нахождения конечной точки титрования определяемого компонента подходящим титрантом, т.е. используют зависимость измеряемого параметра от объема титранта. Для любого рода электрохимических измерений необходима электрохимическая цепь или электрохимическая ячейка, составной частью которой является анализируемый раствор. Электрохимические методы классифицируют в зависимости от типа явлений, замеряемых в процессе анализа. Различают две группы электрохимических методов: 1. Методы без наложения постороннего потенциала, основанные на измерении разности потенциалов, который возникает в электрохимической ячейке, состоящей из электрода и сосуда с исследуемым раствором. Эту группу методов называют потенциометрическими. В потенциометрических методах используют зависимость равновесного потенциала электродов от концентрации ионов, участвующих в электрохимической реакции на электродах. 2. Методы с наложением постороннего потенциала, основанные на измерении: а) Электрической проводимости растворов ̶ кондуктометрия; б) Количества электричества, прошедшего через раствор ̶ кулонометрия; в) Зависимости величины тока от приложенного потенциала ̶ вольт-амперометрия; г) Времени, необходимого для прохождения электрохимической реакции – хроноэлектрохимические методы (хроновольтамперометрия, хронокондуктометрия). В методах этой группы на электроды электрохимической ячейки налагают посторонний потенциал. Основным элементом приборов для электрохимического анализа является электрохимическая ячейка. В методах без наложения постороннего потенциала она представляет собой гальванический элемент, в котором вследствие протекания химических окислительно-восстановительных реакций возникает электрический ток. В ячейке типа гальванического элемента в контакте с анализируемым раствором находятся два электрода – индикаторный электрод, потенциал которого зависит от концентрации вещества, и электрод с постоянным потенциалом – электрод сравнения, относительно которого измеряют потенциал индикаторного электрода. Измерение разности потенциалов производят специальными приборами – потенциометрами [2]. В методах с наложением постороннего потенциала применяют электрохимическую ячейку, названную так потому, что на электродах ячейки под действием наложенного потенциала происходит электролиз – окисление или восстановление вещества. В кондуктометрическом анализе используют кондуктометрическую ячейку, в которой замеряют электрическую проводимость раствора. По способу применения электрохимические методы можно классифицировать на прямые, в которых концентрацию веществ измеряют по показанию прибора, и электрохимическое титрование, где индикацию точки эквивалентности фиксируют с помощью электрохимических измерений. В соответствии с этой классификацией различают потенциометрию и потенциометрическое титрование, кондуктометрию и кондуктометрическое титрование и т.д. Приборы для электрохимических определений кроме электрохимической ячейки, мешалки, нагрузочного сопротивления включают устройства для измерения разности потенциалов, тока, сопротивление раствора, количества электричества. Эти измерения могут осуществляться стрелочными приборами (вольтметр или микроамперметр), осциллографами, автоматическими самопишущими потенциометрами. Если электрический сигнал от ячейки очень слабый, то его усиливают с помощью радиотехнических усилителей. В приборах методов с наложением постороннего потенциала важной частью являются устройства для подачи на ячейку соответствующего потенциала стабилизированного постоянного или переменного тока (зависит от типа метода). Блок электропитания приборов электрохимического анализа включает обычно выпрямитель и стабилизатор напряжения, который обеспечивает постоянство работы прибора. Потенциометрия Потенциометрия основана на измерении разности электрических потенциалов, возникающих между разнородными электродами, опущенными в раствор с определяемым веществом. Электрический потенциал возникает на электродах при прохождении на них окислительно-восстановительной (электрохимической) реакции. Окислительно-восстановительные реакции протекают между окислителем и восстановителем с образованием окислительно-восстановительных пар, потенциал Е которых определяется по уравнению Нернста концентрациями компонентов пар [ок] и [вос]:  , (1) , (1)где Е° – стандартный электродный потенциал, В; n – число электронов, участвующих в процессе. Потенциометрические измерения проводят, опуская в раствор два электрода – индикаторный, реагирующий на концентрацию определяемых ионов, и стандартный электрод или электрод сравнения, относительно которого измеряется потенциал индикаторного. Применяют несколько видов индикаторных и стандартных электродов. Электроды первого рода обратимы относительно ионов металла, из которого состоит электрод. При опускании такого электрода в раствор, содержащий катионы металла, образуется электродная пара: Mn+/M. Электроды второго рода чувствительны к анионам и представляют собой металл М, покрытый слоем нерастворимой его соли МА с анионом A–, к которому чувствителен электрод. При контакте такого электрода с раствором, содержащим указанный анион A–, возникает потенциал Е, величина которого зависит от произведения растворимости соли ПРMAи концентрации аниона [A–] в растворе. Электродами второго рода являются хлорсеребряный и каломельный. Насыщенные хлорсеребряный и каломельный электроды поддерживают постоянный потенциал и применяют в качестве электродов сравнения, по отношению к которым измеряется потенциал индикаторного электрода. Инертные электроды – пластина или проволока, изготовленная из трудноокисляемых металлов – платины, золота, палладия. Применяются они для измерения Е в растворах, содержащих окислительно-восстановительную пару (например, Fe3+/Fe2+). Мембранные электроды различного типа имеют мембрану, на которой возникает мембранный потенциал Е. Величина Е зависит от разности концентраций одного и того же иона по разным сторонам мембраны. Простейшим и наиболее употребляемым мембранным электродом является стеклянный электрод. Смешивание нерастворимых солей типа AgBr, AgCl, AgI и других с некоторыми пластмассами (каучуки, полиэтилен, полистирол) привело к созданию ион-селективных электродов на Br–, Cl–, I–, избирательно адсорбирующих из раствора указанные ионы вследствие правила Панета – Фаянса – Гана. Так как концентрация определяемых ионов вне электрода отличается от таковой внутри электрода, равновесия на поверхностях мембраны отличаются, что приводит к возникновению мембранного потенциала. Для проведения потенциометрических определений собирают электрохимическую ячейку из индикаторного электрода сравнения, который опускают в анализируемый раствор и подсоединяют к потенциометру. Применяемые в потенциометрии электроды имеют большое внутреннее сопротивление (500–1000 МОм), поэтому существуют типы потенциометров представляют собой сложные электронные высокоомные вольтметры. Для измерения ЭДС электродной системы в потенциометрах применяют компенсационную схему, позволяющую уменьшить ток в цепи ячейки. Наиболее часто потенциометры применяют для прямых измерений рН, показатели концентраций других ионов pNa, pK, pNH₄, pCl и мВ. Измерения проводят, используя соответствующие ион-селективные электроды. Для измерения рН применяют стеклянный электрод и электрод сравнения – хлорсеребряный. Перед проведением анализов необходимо проверить калибровку рН-метров по стандартным буферным растворам, фиксаналы которых прикладываются к прибору. рН-метры помимо прямых определений рН, pNa, pK, pNH₄, pCl и других позволяют проводить потенциометрическое титрование определяемого иона. Потенциометрическое титрование Потенциометрическое титрование проводят в тех случаях, когда химические индикаторы использовать нельзя или при отсутствии подходящего индикатора. В потенциометрическом титровании в качестве индикаторов используют электроды потенциометра, опушенные в титруемый раствор. При этом применяют электроды, чувствительные к титруемым ионам. В процессе титрования изменяется концентрация ионов, что регистрируется на шкале измерительного пробора потенциометра. Записав показания потенциометра в единицах рН или мВ, строят график их зависимости от объема титранта (кривую титрования), определяют точку эквивалентности и объем титранта, израсходованный на титрование. По полученным данным строят кривую потенциометрического титрования. Кривая потенциометрического титрования имеет вид, аналогичный кривой титрования в титриметрическом анализе. По кривой титрования определяют точку эквивалентности, которая находится в середине скачка титрования. Для этого проводят касательные к участкам кривой титрования и по середине касательной скачка титрования определяют точку эквивалентности. Наибольшее значение изменения ∆рН/∆V приобретает в точке эквивалентности. Еще более точно точку эквивалентности можно определить методом Грана, по которому строят зависимость ∆V/∆Е от объема титранта. Методом Грана можно проводить потенциометрическое титрование, не доводя его до точки эквивалентности. Потенциометрическое титрование применяют во всех случаях титриметрического анализа. При кислотно-основном титровании используют стеклянный электрод и электрод сравнения. Поскольку стеклянный электрод чувствителен к изменениям рН среды, при их титровании на потенциометре регистрируются изменения рН среды. Кислотно-основное потенциометрическое титрование с успехом применяют при титровании слабых кислот и оснований (рК≤8). При титровании смесей кислот необходимо, чтобы их рК отличались больше, чем на 4 единицы, в противном случае часть более слабой кислоты оттитровывается вместе с сильной, и скачок титрования выражен не четко. Это позволяет использовать потенциометрию для построения экспериментальных кривых титрования, подбор индикаторов для титрования и определения констант кислотности и основности. При осадительном потенциометрическом титровании применяют в качестве индикатора электрод из металла, составляющего с определяемыми ионами электродную пару. При комплексометрическом титровании используют: а) металлический электрод, обратимый к иону определяемого металла; б) платиновый электрод при наличии в растворе окислительно-восстановительной пары. При связывании титрантом одного из компонентов редокс-пары меняется его концентрация, что вызывает изменения потенциала индикаторного платинового электрода. Применяются также обратное титрование избытка раствора ЭДТА, добавленного к соли металла, раствором соли железа (III). При окислительно-восстановительном титровании применяют электрод сравнения и платиновый индикаторный электрод, чувствительный к окислительно-восстановительным парам. Потенциометрическое титрование – один из наиболее употребляемых методов инструментального анализа вследствие простоты, доступности, селективности и широких возможностей. Кондуктометрия. Кондуктометрическое титрование Кондуктометрия основана на измерении электрической проводимости раствора. Если в раствор вещества поместить два электрода и подать на электроды разность потенциалов, то через раствор потечет электрический ток. Как и каждый проводник электричества, растворы характеризуются сопротивлением R и обратной ему величиной – электрической проводимостью L: 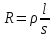 , (2) , (2)где R – сопротивление, Ом;  – удельное сопротивление, Ом . см; – удельное сопротивление, Ом . см;S– площадь поверхности, см2.  , (3) , (3)где L– электрическая проводимость, Ом–1; R – сопротивление, Ом. Кондуктометрический анализ проводят с помощью кондуктометров – приборов, измеряющих сопротивление растворов. По величине сопротивления R определяют обратную ему по величине электрическую проводимость растворов L. Определение концентрации растворов осуществляют прямой кондуктометрией и кондуктометрическим титрованием. Прямая кондуктометрия используется для определения концентрации раствора по калибровочному графику. Для составления калибровочного графика замеряют электрическую проводимость серии растворов с известной концентрацией и строят калибровочный график зависимости электрической проводимости от концентрации. Затем измеряют электрическую проводимость анализируемого раствора и по графику определяют его концентрацию. Чаще применяют кондуктометрическое титрование. При этом в ячейку с электродами помещают анализируемый раствор, ячейку помещают на магнитную мешалку и титруют соответствующим титрантом. Титрант добавляют равными порциями. После добавления каждой порции титранта замеряют электрическую проводимость раствора и строят график зависимости между электрической проводимостью и объемом титранта. При добавлении титранта происходит изменение электрической проводимости раствора в т.э. наступает перегиб кривой титрования. От подвижности ионов зависит электрическая проводимость раствора: чем выше подвижность ионов, тем больше электрическая проводимость раствора. Кондуктометрическое титрование обладает рядом преимуществ. Его можно проводить в мутных и окрашенных средах, в отсутствии химических индикаторов. Метод обладает повышенной чувствительностью и позволяет анализировать разбавленные растворы веществ (до 10–4 моль/дм³). Кондуктометрическим титрованием анализируют смеси веществ, т.к. различия в подвижности различных ионов существенны и их можно дифференцированно оттитровывать в присутствии друг друга. Кондуктометрический анализ легко автоматизировать, если раствор титранта подавать из бюретки с постоянной скоростью, а изменение электрической проводимости раствора регистрировать на самописце. Эта разновидность кондуктометрии получила название хроно-кондуктометрического анализа. В кислотно-основном титровании кондуктометрическим путем можно определять сильные кислоты, слабые кислоты, соли слабых оснований и сильных кислот. В осадительном кондуктометрическом титровании электрическая проводимость титруемых растворов сначала уменьшается или остается на некотором постоянном уровне вследствие связывания титруемого электролита в осадок, после т.э. при появлении избытка титранта – снова возрастает. В комплексометрическом кондуктометрическом титровании изменения электрической проводимости раствора наступают вследствие связывания катионов металла в комплекс с ЭДТА. Окислительно-восстановительное кондуктометрическое титро-вание основано на изменении концентрации реагирующих ионов и появлении в растворе новых ионов, что изменяет электрическую проводимость раствора. В последние годы получило развитие высокочастотная кондуктометрия, в которой электроды с раствором не контактируют, что важно при анализе агрессивных сред и растворов в закрытых сосудах. Получила развитие два варианта – прямая высокочастотная кондуктометрия и высокочастотное титрование. Прямая высокочастотная кондуктометрия применяется для определения влажности веществ, зерна, древесины, концентрации растворов в закрытых сосудах – ампулах, при анализе агрессивных жидкостей. Высокочастотное титрование проводят на специальных титраторах – ТВ-6, ТВ-6Л. Высокочастотное кондуктометрическое титрование проводят по типу кислотно-основного, окислительно-восстановительного или осадительного титрования в тех случаях, когда отсутствует подходящий индикатор или при анализе смесей веществ. Кулонометрия. Кулонометрическое титрование В кулонометрии вещества определяют измерением количества электричества, затраченное на их количественное электрохимическое превращение. Кулометрический анализ проводят в электролитической ячейке, в которую помещают раствор определяемого вещества. При подаче на электроды ячейки соответствующего потенциала происходит электрохимическое восстановление или окисление вещества. Согласно законам электролиза, открытым Фарадеем, количество вещества, прореагировавшего на электроде, пропорционально количеству электричества, прошедшего через раствор:  , (4) , (4)где g – масса, выделяющегося вещества, г; n – количество электронов, переносимых в электродном процессе; F – число Фарадея (F = 96485 Кл/моль); I – сила тока, А; t – время, с; M – молярная масса выделяющегося вещества, г/моль. Кулонометрический анализ позволяет определять вещества, не осаждающиеся на электродах или улетучивающиеся в атмосферу при электрохимической реакции. Различают кулонометрию прямую и кулонометрическое титрование. Высока точность и чувствительность методов измерения электрического тока обеспечивает кулонометрическому анализу уникальную точность 0,1-0,001%, и чувствительность до 1∙10-8 ̶ 1∙10-10 г. Поэтому кулонометрический анализ применяется для определения микропримесей и продуктов разрушения веществ, что важно при контроле их качества. Для индикации т.э. при кулонометрическом титровании можно применять химический и инструментальные методы – добавление индикаторов, обнаружение окрашенных соединений фотометрическим или спектрофотометрическим путём. В отличии от других методов анализа кулонометрия может быть полностью автоматизирована, что сводит к минимуму случайные ошибки определения. Эта особенность использована при создании автоматических кулонометрических титраторов – чувствительных приборов, применяющихся для особо точных анализов, когда другие методы оказываются недостаточно чувствительными. При анализе веществ, малорастворимых в воде, кулонометрию можно проводить на электродах из ацетиленовой сажи, являющиеся хорошим адсорбентом и извлекающий такие вещества из реакционной среды с достаточной полнотой. Кулонометрическое титрование – перспективный метод инструментального анализа. Он может найти широкое применение для решения ряда специальных аналитических задач – анализа примесей, малых количеств лекарственных препаратов, определение в биологическом материале и окружающей среде токсических веществ, микроэлементов и других соединений [3]. |