современные химические методы анализа. Тема 1 Методы аналитической химии
 Скачать 465.58 Kb. Скачать 465.58 Kb.
|

|
Тема № 5 Кинетические методы. Сущность кинетических методов, их место среди других методов анализа. Скорость химических реакций имеет большое значение в аналитической химии. Измерение скорости реакций лежит в основе кинетических методов анализа. Медленное протекание некоторых реакций затрудняет анализ. Иногда, наоборот, небольшая скорость химических процессов благоприятствует выполнению анализа и играет положительную роль. Исследование скорости реакций – предмет химической кинетики. Скорость реакций определяется количеством веществ, прореагировавших за единицу времени. Рассмотрим реакцию между веществами А и В: A + B = C + D. Ее скорость зависит от концентрации реагирующих компонентов: V = dx/dt = K [A] [B], (1) где х – количество вступившего в реакцию [A] (или [B] ) за время t; К – константа скорости реакции. В самом деле, при увеличении концентрации возрастает общее количество частиц в единице объема, число их столкновений между собой и, следовательно, вероятность взаимодействия; поэтому за единицу времени прореагирует больше вещества, чем при малых концентрациях. Физический смысл константы скорости выясняется, если принять концентрации [A] и[B] равными 1 моль/л. Тогда V = dx/dt = K, (2) т.е. константа скорости равна количеству вещества, прореагирующего за единицу времени при [A] = [B] = 1 моль. Скорость реакций может изменяться в очень широких пределах – от малых долей секунды до нескольких часов или дней. Условно все реакции обычно делят на быстрые и медленные. К быстрым реакциям относят такие, в которых половина имеющегося количества вещества реагирует за 10 с или за меньшее время, все остальные реакции относят к медленным. Очень быстрые и очень медленные реакции в химико- аналитических целях являются малопригодными. Хотя трудно установить какие-либо жесткие правила или критерии, ограничивающие применение тех или иных реакций в кинетических методах анализа, все же некоторые пределы применимости можно отметить. Почти повсеместно принято считать, что аналитическая реакция ( в кинетических методах ее часто называют индикаторной реакцией) должна продолжаться не менее 1 мин и не более 2 ч. Более быстрые реакции, как правило, не применяются потому, что при использовании обычного обычного оборудования химико –аналитической лаборатории скорость таких реакций трудно измерить с достаточной точностью. При наличии специального оборудования эти затруднения отпадают и соответствующее ограничение снимается. Реакции, продолжающиеся более 2 ч, нежелательны из-за большой длительности анализа. Оптимальным временем для измерения скорости реакции считается 10 – 15 мин. Указанные пределы в значительной степени условны, поскольку можно в достаточно широких пределах регулировать скорость химической реакции, например, изменением температуры, концентрации реагирующих веществ или введением в раствор катализаторов ( или ингибиторов). Примером быстрых реакций могут служить многие реакции между ионами противоположного знака заряда, например реакции между водородными и гидроксильными ионами, между окислителями и восстановителями и т.д. Однако существует и очень медленные реакции. Так, исходя из значения стандартных потенциалов, следовало ожидать, что должна происходить реакция между перманганатом и водой с выделением кислорода. Но эта реакция происходит с очень малой скоростью, в связи с чем водные растворы перманганата калия вполне устойчивы и не изменяют своей концентрации длительное время. Известны и другие реакции, характеризующиеся большими значениями констант равновесия, но протекающие с очень малой скоростью. Наоборот, реакции с меньшими константами часто протекают быстро. Примером могут служить следующие реакции: I2 + AsO2 - + 2H2 O = 2I- + AsO4 3- + 4H+ , (3) H2 O2 + AsO2 - = AsO4 3- + 2H+ . (4) Реакция (3) – быстрая, а реакция (4) – очень медленная. В то же время окислительный потенциал иода значительно меньше, чем у пероксида водорода, и, следовательно, константа равновесия реакции (3) меньше, чем реакции (4). Значения констант равновесия можно найти по уравнению: lg K = (E01 – E02 )n / 0,058. (5) Имея в виду, что Е0 ( I2 /2I- ) = 0,62 B, E0 (H2 O2 /H2 O) = 1,77 B и E0 (AsO4 3- /AsO2 - ) = 0,56 В, для реакций (3) и (4) получаем соответственно: lg K3 = 2,1 lg K4 = 41,7, т.е. в термодинамическом смысле реакция (4) значительно более вероятна, чем реакция (3), и должна быть сдвинута в правую сторону в большей степени, однако скорости обеих реакций находятся в обратно пропорциональном отношении к значениям констант равновесия. Пользуясь различными скоростями реакций, нередко удается определять одно вещество в присутствии другого даже в том случае, если реагент взаимодействует с обоими веществами. Рассмотрим две реакции: 2Ce(SO4 )2 + HAsO2 + 2H2 O = Ce2 (SO4 )3 + H3 AsO4 + H2 SO4 , (6) 2Ce(SO4 )2 + 2FeSO4 = Ce2 (SO4 )3 + Fe2 (SO4 )3 (7) сульфат церия окисляет и мышьяк, и железо (II). Однако реакция (6) идет с очень малой скоростью, в то время как реакция (7) происходит мгновенно. Таким способом удается оттитровать железо в присутствии мышьяка. Затем прибавляют катализатор OsO4 , который ускоряет реакцию (6), и оттитровывают мышьяк. Другой пример – титрование иода тиосульфатом в растворе, содержащем свободную кислоту. Тиосульфат взаимодействует и с иодом, и с кислотой: I2 + 2Na2 S2 O3 = 2NaI + Na2 S4 O6 , (8) 2HCl + Na2 S2 O3 = H2 SO3 + 2NaCl. (9) правильные результаты определения иода по реакции (8) можно получить только потому, что скорости обеих реакций различаются во много раз. Реакция (9) протекает настолько медленно, что в процессе восстановления иода тиосульфат не успевает разложиться в соответствии с уравнением (9). Скорость реакции можно регулировать различными способами. Из уравнения (1) видно, что скорость зависит от концентрации реагирующих веществ; следовательно, быстрая реакция замедляется при уменьшении концентрации А и В, и, наоборот, она возрастает, если концентрации соответствующих веществ увеличиваются. Скорость реакций зависит от температуры. Зависимость константы скорости реакции от температуры определяется уравнением Аррениуса d ln K/dT = E/RT2 , (10) где К – константа скорости; Е – энергия активации. В случае простых реакций, идущих в одну стадию, параметр Е показывает, какой минимальной энергией ( в расчете на 1 моль) должны обладать реагирующие частицы, чтобы они могли вступить в химическую реакцию. Частицы, энергия которых больше или равна Е, называют активными. В среднем повышение температуры на 100 С приводит к увеличению скорости реакций в растворе приблизительно в 2-3 раза. Этот прием часто используют в анализе. Так, реакция меду щавелевой кислотой и перманганатом в холодных растворах идет с очень маленькой скоростью, однако нагревание до 80 – 900 С значительно ускоряет реакцию. Растворение металлов или их солей идет значительно быстрее при нагревании. При осаждении малорастворимых соединений нагревание раствора способствует увеличению скорости движения ионов в растворе и приводит к быстрому росту центров кристаллизации и вследствие этого к образованию крупнокристаллических осадков. В кинетических и каталитических методах анализа нередко необходимо в определенный момент времени замедлить или вообще остановить реакцию – охлаждение раствора является одним из методов такого замедления. На скорость реакций влияет характер растворителя. Имеет значение диэлектрическая проницаемость растворителя. Для наиболее распространенного в аналитической химии случая, когда реагируют между собой ионы противоположного знака, скорость реакции уменьшается с увеличением диэлектрической проницаемости. Большинство органических растворителей имеет диэлектрические проницаемости меньше, чем у воды, и поэтому скорость реакций в таких растворителях больше, чем в водных растворах. Однако такая корреляция наблюдается далеко не всегда. Она справедлива только в пределах группы растворителей одного гомологического ряда или для серии смесей переменного состава, приготовленных из определенной пары растворителей. Существенное влияние на скорость химических реакций имеет сольватация реагентов и активированного комплекса. Сольватация последнего понижает его энергию, что в конечном счете ускоряет химическую реакцию. Растворители по их сольватирующей способности можно классифицировать следующим образом: 1) протонные растворители – легко отщепляют протон и обычно содержат группы –ОН, =NH, их отличительная особенность – способность к образованию водородной связи. Типичные растворители этого типа: вода, спирты, карбоновые кислоты, фенолы, аммиак; 2) апротонные растворители - не имеют кислотного водорода, они хорошо сольватируют катионы, практически не сольватируя анионы. Типичные растворители этого типа : ацетон, сульфолан, диметилформамид, диэтиловый эфир, диоксан; 3) инертные растворители характеризуются малой диэлектрической проницаемостью и малым дипольным моментом. К таким растворителям относятся алканы, алкены, ароматические углеводороды, четыреххлористый углерод, сероуглерод. Эти растворители обладают малой сольватирующей активностью, ускоряя химические реакции в основном за счет гомогенизации реакционной среды; 4) растворители с электрофильными свойствами представлены сильными кислотами или суперкислотными системами, например, FSO3 H, CF3 COOH, FSO3 H/SbF5 , кислоты Льюиса. Скорость реакций зависит от ионной силы раствора. В наиболее простом случае, когда взаимодействуют ионы А и В в растворе, зависимость логарифма константы скорости К от ионной силы раствора выражается следующим уравнением: lg K = lg K0 + aZA ZB √μ, (11) где K0 – константа скорости реакции в среде, для которой коэффициенты активности исходных частиц А и В и активированного комплекса между ними приняты равными единице; а – константа, включающая величины ддиэлектрической проницаемости растворителя и абсолютной температуры; Za и ZB – заряды частиц; μ – ионная сила раствора. Из уравнения видно, что с увеличением ионной силы, т.е. с введением в реакционную систему посторонних хорошо диссоциирующих солей, скорость реакции между ионами противоположного знака заряда уменьшается. Объяснение заключается в том, что ионы посторонних солей образуют вокруг реагирующих ионов ионную атмосферу из ионов противоположного знака заряда, которая препятствует непосредственному контакту между ионами и на разрушение которой необходимо затратить определенное время. Наоборот, если реагируют между собой ионы с зарядами одинакового знака, скорость реакции должна возрастать. Изменение концентрации ионов водорода в рекциях окисления – воостановления влияет не только на окислительный потенциал системы, в соответствии с уравнением Нернста, но также нередко на скорость реакции. Так, окислительные потенциалы систем Fe3+ /Fe2+ и I2 /2I- не изменяются в зависимости от рН, однако скорость реакции 2Fe3+ + 2I- = 2Fe2+ +I2 (12) сильно возрастает с увеличением концентрации водородных ионов раствора. Аналогичное влияние на скорость процесса оказывает рН раствора реакции 2Cu2+ + 4I- = Cu2 I2 + I2 (13) использующейся для иодометрического определения меди. Видно, что ионы водорода не участвуют в суммарном уравнении реакции и поэтому не влияют на окислительный потенциал реагирующих веществ. Однако при слишком малой кислотности раствора реакция (13) проходит очень медленно и ее скорость возрастает с уменьшением рН раствора. Скорость реакций зависит от введения катализаторов. Катализаторы могут ускорять или замедлять реакции, в последнем случае говорят об ингибирующем действии. Ускорение реакций связано с тем, что катализатор, взаимодействуя с реагентами, образует неустойчивые промежуточные активные соединения (одно или несколько), понижая энергию активированных комплексов (одного или нескольких). Природа такого взаимодействия зависит от типа катализа. В реакциях имеют место гомогенный, гетерогенный, ферментативный катализы. Методы, основанные на каталитическом действии вещества, представляют собой разновидность кинетических методов, их называют каталитическими. Классификация кинетических методов. Каталитический и некаталитический варианты методы. Кинетические методы основаны на использовании зависимости скорости химической реакции от концентрации реагирующих веществ, а в случае каталитических реакций и от концентрации катализатора: где v — скорость; K — константа скорости каталитической реакции; сА, сВ и скат — концентрации реагирующих веществ и катализатора; m, n и р — показатели степени при концентрациях реагентов и катализатора (обычно р = 1). Аналитическим сигналом в кинетических методах является скорость процесса или пропорциональная ей величина. Реакцию, скорость которой измеряется, называют индикаторной, а вещество, по изменению концентрации которого судят о скорости процесса — индикаторным веществом. Индикаторные реакции могут быть основаны на катализе окислительно-восстановительных реакций, реакций замещения в координационной сфере ионов металлов, реакций гидролиза и декарбоксилирования органических соединений. Наиболее чувствительны и сравнительно просто выполнимы окислительно-восстановительные каталитические реакции. Они чаще всего используются в анализе неорганических веществ. Кроме каталитических реакций в кинетических методах используют и некаталитические реакции окисления-восстановления, разложения, осаждения. К индикаторной реакции предъявляют ряд требований: концентрация определяемого компонента за время наблюдения практически не должна меняться. Катализатор в ходе реакции не расходуется. Если же определяемым является одно из реагирующих веществ (некаталитический вариант метода), то с достаточной точностью его можно определять в тот начальный период, когда его концентрация изменяется не более чем на 5%; необходимо наличие быстрого, простого и доступного метода наблюдения за скоростью индикаторной реакции, т. е. за изменением концентрации индикаторного вещества во времени. скорость индикаторной реакции должна находиться в определенных пределах. Оптимальное время наблюдения за скоростью индикаторной реакции 5–15 мин. Однако с развитием методов изучения быстрых процессов все чаще используют реакции, протекающие с большой скоростью. Существуют два варианта кинетических методов. В каталитическом варианте кинетического метода (каталитическом методе, каталиметрии) определяемый компонент или связанные с ним соединения являются катализатором индикаторной реакции. В некаталитическом варианте кинетического метода определяемым компонентом является одно из реагирующих веществ в некаталитической или каталитической индикаторной реакции. Каталитические методы отличаются от других химических методов анализа высокой чувствительностью, а некаталитический вариант кинетических методов – высокой селективностью. Возможны различные способы определения неизвестной концентрации вещества по данным кинетических измерений. Если индикаторным веществом является продукт реакции, и его текущую концентрацию обозначить через х, то скорость реакции можно выразить как 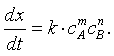 (13.2) (13.2)На начальной стадии реакции концентрации определяемого вещества В и реагента А могут практически не изменяться за время наблюдения за скоростью процесса. Тогда, проинтегрировав уравнение (2), получим т. е. наблюдается линейная зависимость между концентрацией индикаторного вещества и временем. Кинетический метод, основанный на использовании этого уравнения, называют дифференциальным. Если концентрация хотя бы одного из реагирующих веществ за время наблюдения за скоростью реакции заметно меняется (более чем на 10%), то между концентрацией индикаторного вещества и временем существует более сложная (например, логарифмическая, обратная и т. д.) зависимость. Такой кинетический метод называют интегральным. В интегральном варианте часто применяют построение зависимостей концентрации индикаторного вещества от времени в полулогарифмических, обратных или других координатах, т. к. для расчета неизвестной концентрации определяемого компонента удобнее использовать прямоугольные участки кинетических кривых. Характер кинетических кривых, а следовательно, и использование дифференциального или интегрального вариантов кинетических методов анализа определяется типом индикаторной реакции, ее механизмом. В настоящее время наиболее распространенными являются три основных способа определения неизвестной концентрации по данным кинетических измерений [1]. Это способы тангенсов, фиксированного времени, фиксированной концентрации. Рассмотрим их применительно к дифференциальному варианту кинетического метода анализа. Способ тангенсов основан на определении тангенса угла наклона кинетических кривых tga при известных концентрациях определяемого вещества. При этом tga характеризует скорость индикаторной реакции и зависит от концентрации определяемого вещества. Градуировочный график строят в координатах: концентрация определяемого соединения — tga (рис. 13.1, а). Способ фиксированного времени. При определенном, строго фиксированном интервале времени протекания реакции, измеряют концентрацию индикаторного вещества в пробах с известными концентрациями определяемого компонента. Градуировочный график строят в координатах концентрация определяемого вещества — концентрация индикаторного вещества при фиксированном времени протекания реакции tфикс. (рис. 13.1, б). Часто при работе этим методом индикаторную реакцию останавливают при tфикс.. Путем резкого охлаждения, изменения кислотности раствора, добавления ингибиторов. Способ фиксированной концентрации. В отдельных пробах с известными концентрациями определяемого вещества проводят индикаторную реакцию до строго определенной (фиксированной) концентрации индикаторного вещества хфикс. и измеряют время достижения этой концентрации. Градуировочный график строят в координатах: концентрация определяемого компонента — величина, обратная времени достижения хфикс. (рис. 13.1, в). В интегральном варианте все способы определения неизвестной концентрации аналогичны, лишь между временем реакции и концентрацией индикаторного вещества существует более сложная функциональная зависимость. В этом случае находят функции концентрации индикаторного вещества, линейно изменяющиеся во времени (логарифмическая, обратная и т. д.). 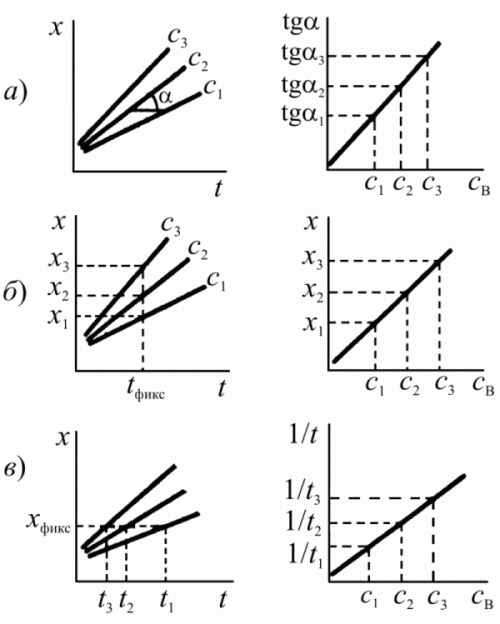 Рис. 13.1. Способы определения неизвестной концентрации по данным кинетических измерений: а — тангенсов; б — фиксированного времени; в — фиксированной концентрации (х — концентрации индикаторного вещества, t — время, с3 > c2 > c1 — концентрации определяемого соединения В) Каталиметрическое титрование — процесс титрования в присутствии катализатора, в котором точку конца титрования определяют по резкому увеличению или уменьшению скорости реакции. С целью автоматизации каталиметрического метода анализа скорость реакции часто измеряют в открытых системах. Открытой называют систему, в которую по мере протекания реакции вводят реагенты и/или из которой отводят продукты реакции. В ходе реакции растворы подаются в реакционную камеру с постоянной или регулируемой скоростью. Разработаны разные варианты открытых систем: на основе проточных методов и «стат»-методов. Проточные методы. К ним относится метод непрерывной струи, основанный на смешении реагентов в струе и предложенный для быстро протекающих реакций с периодом полупревращения t1/2 = 0,01–10 с. Другой вариант проточного метода применяют для измерения скоростей сравнительно медленно протекающих реакций с t1/2 = 1–10 мин. В этом случае проточная ячейка одновременно является и смесительной камерой. Исходные реагенты индикаторной реакции и анализируемый раствор, содержащий катализатор с концентрацией скат, непрерывно подаются насосами в смесительную камеру вместимостью около 10 мл, продукты реакции и реагенты вытекают со скоростью 2–20 мл/мин. При каждом значении скат устанавливается постоянная концентрация индикаторного вещества и фиксируется постоянный сигнал, соответствующий скат. Смена раствора в кювете происходит за 1–2 мин, что определяет производительность анализатора 30 проб в час. Стат-метод предполагает введение реагентов со скоростью, равной скорости их расходования в реакции, так что концентрация индикаторного вещества остается постоянной. Скорость введения реагента регулируется автоматически. Воспроизводимость результатов кинетических измерений повышается при использовании метода одновременного компарирования. В анализируемый раствор и растворы шкалы стандартов одновременно с помощью стартовой пипетки вводят реагент, инициирующий протекание каталитической реакции. Через определенный промежуток времени сравнивают аналитические сигналы анализируемого раствора и шкалы стандартов и оценивают содержание определяемого вещества. Метод не требует термостатирования. Для учета влияния примесей на скорость реакции применяют метод добавок. Скорость реакции измеряют в равных аликвотных частях анализируемого раствора без добавки и в присутствии определенных добавок катализатора. Метод добавок дает правильные результаты, если в растворе отсутствуют посторонние примеси, обладающие каталитическим действием на индикаторную реакцию. Скорость реакции можно определять по времени внезапного появления окраски раствора в реакциях Ландольта. Реакции Ландольта — это медленные химические реакции, в которых образование окрашенного продукта реакции задерживается подходящим реагентом, специально добавленным для этой цели. Например, при окислении бромида персульфатом, катализируемом ионами меди(II), образующийся бром окисляет аскорбиновую кислоту и не взаимодействует с индикатором N,N-диметил-п-фенилендиамином. Когда практически вся аскорбиновая кислота окислится, появляется окраска индикатора. Метод, основанный на эффекте Ландольта, в ряде случаев обеспечивает более высокую воспроизводимость результатов анализа, чем обычный метод фиксированной концентрации, разновидностью которого он является. Концентрацию катализатора можно определять по длительности индукционного периода tинд., по истечении которого скорость реакции становится заметной (рис. 13.2). Этот способ является разновидностью метода фиксированной концентрации. Индукционный период наблюдается не только в реакциях Ландольта, но и в автокаталитических реакциях, а также в реакциях, когда в начальный период изменяется соотношение форм катализатора. Длительность индукционного периода связана с концентрацией катализатора зависимостью 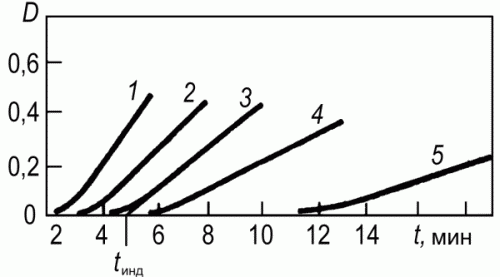 Рис. 13.2. Кинетические кривые окисления KI (2 × 10–4 М) пероксидом водорода (2,4 × 10–3 М), катализируемого Ti(IV), в присутствии крахмала За изменением концентрации индикаторного вещества во времени можно наблюдать любым методом, и при построении кинетических кривых вместо концентрации образующегося продукта использовать любую, пропорциональную ей величину — оптическую плотность, силу тока, потенциал системы и т. д. Чаще всего для наблюдения за скоростью индикаторной реакции используют спектрофотометрические и люминесцентные, реже — электрохимические, термометрические и титриметрические методы. |