учебник по патфизу. Учебник для слушателей и курсантов военномедицинской академии и военномедицинских институтов под редакцией проф. В. Ю. Шанина
 Скачать 4.96 Mb. Скачать 4.96 Mb.
|

|
Классификация гипоксических состояний ГИПОКСИЯ, возникающая в результате возмущающих воздействий на входе функциональной системы, ответственной за кислородное обеспечение организма:
ГИПОКСИЯ, возникающая в результате изменений в отдельных звеньях функциональной системы, ответственной за кислородное обеспечение организма:
ГИПОКСИЯ НАГРУЗКИ, возникающая в результате увеличения нагрузки на функциональную систему, ответственную за кислородное обеспечение организма. По времени развития гипоксия классифицируется:
По характеру развития:
Этиология и патогенез отдельных форм гипоксии Гипоксический тип гипоксии (экзогенная гипоксия) развивается в результате снижения рО2 во вдыхаемом воздухе. Наиболее типичным ее проявлением являются горная и высотная болезни. Гипоксическая гипоксия может возникнуть во всех случаях, когда осуществляется дыхание газовыми смесями с недостаточным парциальным давлением кислорода. Необходимо помнить, что гипоксическая гипоксия может возникнуть при дыхании в замкнутом пространстве (отсеки подводной лодки, хранилища, бункера, ангары), а также при неисправности дыхательной аппаратуры. При гипоксической гипоксии рО2 снижается как в альвеолярном воздухе, так и в артериальной крови, тканях. Уменьшается общий венозно-воздушный градиент. Выделяют 4 степени тяжести гипоксии в зависимости от рО2 артериальной крови:
Из приведенных данных видно, что летальным считается рО2, соответствующее нескольким десяткам мм рт. ст., то есть когда содержание кислорода во вдыхаемом воздухе 3 уменьшается на 60% и более. Одной из распространенных форм гипоксической гипоксии является высотная болезнь - остро развивающееся состояние, в котором выделяют 2 формы:
Горная болезнь развивается при пребывании в условиях высокогорья или при длительном нахождении в барокамере в условиях гипобарии. Помимо парциального давления кислорода в механизмах развития горной болезни существенное значение имеют влажность воздуха, инсоляция, сильные ветры, низкая минерализация питьевой воды (см. таблицу 5.1). Таблица 5.1 Группировка компонентов горного комплекса по постоянству воздействия на человека
Поэтому течение горной болезни отличается на одних и тех же высотах, но в разной местности. Выделяют следующие формы горной болезни:
По длительности течения выделяют:
Основной этиологический фактор горной болезни – это снижение парциального давления кислорода в альвеолярной газовой смеси, обусловленное низким парциальным давлением кислорода во вдыхаемой газовой смеси. От горной болезни страдают 30% неадатированных к высотной гипоксемии людей после быстрого подъема на высоту большую, чем 3000 м над уровнем моря. У 75% неприспоболенных субъектов симптомы острой горной болезни выявляют после быстрого подъема на высоту, превышающую 4500 м над уровнем моря. Головная боль как первый признак начала развития горной болезни связана со спазмом сосудов головного мозга в ответ на падение напряжения углекислого газа в артериальной крови в результате компенсаторной гипервентиляции, обусловливающей гипокапнию, но не устраняющей артериальной гипоксемии. Когда напряжение кислорода в артериальной крови не больше, чем 60 мм рт.ст., то значительный гипоэргоз церебральных нейронов, несмотря противодействие системы ауторегуляции локальной скорости мозгового кровотока, обусловливает расширение артериол и раскрытие прекапилярных сфинктеров в системе микроциркуляции мозга. В результате увеличивается кровоснабжение головного мозга, что повышает внутричерепное давление и проявляет себя головной болью. Компенсаторная гипервентиляция у страдающих горной болезнью на высотах в диапазоне 3000-4500 м над уровнем моря вызывает респираторный алкалоз и бикарбонатурию как компенсаторную реакцию на снижение содержания протонов и рост бикарбонатного аниона во внеклеточной жидкости и клетках. Бикарбонатурия усиливает натрийурез и, снижая содержание в организме натрия, уменьшает объем внеклеточной жидкости и даже обусловливает гиповолемию. При подъеме на высоты, на которых компенсаторные реакции в ответ на гипоксическую гипоксию не в состоянии предотвратить связанного с ней гипоэргоза клеток, гипервентиляция через повышение потребления кислорода организмом обостряет системный гипоэргоз. Усиление системного гиперэргоза на уровне всего организма повышает интенсивность анаэробного гликолиза, что вызывает метаболический лактатный ацидоз типа А. Патологически низкое парциальное давление кислорода во вдыхаемой газовой смеси служит стимулом для «альвеоло-капиллярного рефлекса» с еще не выявленным центральным звеном. В эфферентном звене, на уровне эффектора, рефлекс сужает легочные венулы и артериолы, что обусловливает легочную первичную как венозную, так и артериальную гипертензию. Легочная артериальная гипертензия может приводить к острой правожелудочковой недостаточности в результате патогенно высокой постнагрузки правого желудочка. Полагают, что легочная венозная гипертензия и отрицательные нервные влияния на легочную паренхиму в ответ на гипоксическую гипоксию обуславливают некардиогенный отек легких как осложнение горной болезни. При этом отрицательные нервные влияния на легкие в частности приводят к росту экспрессии флогогенного потенциала клеточных эффекторов воспаления, локализованных в легких, то есть эндотелиоцитов, нейтрофилов, тучных клеток и клеток мононуклеарных фагоцитов (не имеющее биологического смысла нейрогенное воспаление). Гипербарическая гипоксия развивается в условиях повышенного парциального давления воздуха (например, при дыхании в барокамере). Известно, что повышение давления вдыхаемого воздуха на каждую атмосферу при неизменной температуре влечет за собой дополнительное растворение в 100 мл крови 2,3 мл кислорода (закон Генри-Дальтона). Высокое парциальное давление кислорода во вдыхаемом воздухе приводит к увеличению его напряжения в артериальной крови и тканях. Таким образом, основным звеном патогенеза гипербарической гипоксии является повышение напряжения кислорода в тканях организма. Гипероксическая гипоксия возникает вследствие патогенно высокого парциального давления кислорода во вдыхаемой смеси газов, которое обуславливают: а) рост содержания кислорода во вдыхаемой газовой смеси; б) увеличение давления (барометрического, атмосферного) смеси газов. Гипероксическая гипоксия - возникает при дыхании газовыми смесями, содержащими кислорода больше, чем во вдыхаемом атмосферном воздухе (при нормальном атмосферном давлении –760 мм рт. ст., парциальное давление кислорода в окружающем нас воздухе равно 159 мм рт.ст.). Основным звеном патогенеза развивающимся в данном случае является повышение напряжения кислорода в тканях. Гипероксическая гипоксия - это следствие токсичного действия кислорода при его аномально высоких парциальном давлении в альвеолярной газовой смеси и напряжении в артериальной крови и в тканях. Под токсичным действием кислорода понимают повреждения тканей, клеток и интерстициальных тканевых структур, обусловленные свободнорадикальным окислением. Токсическому действию кислорода особенно подвержены старики, у которых со старением падает активность антиоксидантных систем, в частности ферментов супероксиддисмутазы, каталаз и пероксидазы. Циркуляторная гипоксия при травматическом шоке предрасполагает к токсическому эффекту кислорода, вызывая через свободнорадикалыюе окисление и дефицит свободной энергии недостаточность антиоксидантных систем. При гипероксической гипоксии высокое напряжение кислорода в тканях ведет к окислительной деструкции внутриклеточных митохондриальных структур, что угнетает тканевое дыхание или снижает эффективность улавливания клеткой свободной энергии при биологическомокислении. Патогенез гипероксической гипоксии весьма сложен, так как этот вид кислородной недостаточности сопровождается разнообразными сдвигами обмена веществ под влиянием высокого напряжения кислорода в тканях. Прежде всего происходит инактивация многих энзимов, особенно тех, что содержат сульфгидрильные группы. Одним из следствий системной ферментопатии при гипероксической гипоксии выступает падение содержания в мозге гамма-аминобутирата, главного тормозного медиатора серого вещества, что обуславливает судорожный синдром кортикального генеза. Высокое напряжение кислорода в тканях приводят к усиленному образованию свободных кислородных радикалов, нарушающих образование дезоксирибонуклеиновой кислоты, и тем самым извращающих внутриклеточный синтез белка. Кроме того, свободнорадикальное окисление фосфолипидов клеточных мембран как фактор первичной альтерации тканей служит инициирующим моментом воспаления. При увеличении парциального давления кислорода во вдыхаемой газовой смеси в первую очередь патологические изменения возникают в легочной паренхиме, в которой в наибольшей степени возрастают напряжение кислорода и образование свободных кислородных радикалов. Токсический эффект кислорода может клинически значимо проявить себя уже при возрастании парциального давления кислорода во вдыхаемой газовой смеси до 200 мм рт. ст., если больной непрерывно дышит такой смесью в течение нескольких часов. При парциальных давлениях кислорода во вдыхаемой смеси газов меньших, чем 736 мм рт. ст., гистотоксический эффект высокого напряжения кислорода в тканях приводит в основном к воспалительным изменениям легочной ткани, а у некоторых больных и к некардиогенному отеку легких. Кроме того, у больных выявляют диффузное микроателектазирование легких из-за разрушения свободнорадикальным окислением системы сурфактанта. Дыхание газовой смесью, парциальное давление кислорода в которой выше, чем 4416 мм рт. ст., приводит к тоникоклоническим судорогам и потере сознания в течение нескольких минут. Резистентность по отношению к гистотоксическому эффекту высокого напряжения кислорода в тканях снижает наследственная недостаточность антиоксидантных систем клетки, в частности низкая активность ферментов глютатионпероксидазы и супероксиддисмутазы. Высокие парциальное давление кислорода в альвеолах и его напряжений в артериальной крови и тканях являются патогенными раздражителями, которые вызывают дисфункции элементов респиронов. В основном эти расстройства складываются из нарушений легочной микроциркуляции, обусловленных спазмом микрососудов в ответ на избыточные нервные регуляторные влияния. Это индуцирует патологическую вариабельность вентиляционно-перфузионных отношений структурно-функциональных элементов легких, а через активацию эндотелия легочных микрососудов предположительно предрасполагает к воспалению лишенному биологической цели, вторичная альтерация при котором разрушает респироны. Таким образом, рассматривая механизм гипероксической и гипербарической гипоксии, необходимо подробно охарактеризовать токсическое действие кислорода. Кислородная интоксикация проявляется в двух формах:
Острая интоксикация возникает обычно при сравнительно кратковременной экспозиции кислорода под давлением более 3 атм. Хроническое кислородное отравление развивается, как правило, при длительном воздействии малых избыточных давлений кислорода, а также при нормобарической гипероксии. У взрослых людей при дыхании чистым кислородом под обычным давлением (760 мм рт. ст.) через 6-12 часов появляются болезненные ощущения в груди, кашель, боли в горле. Дальнейшая экспозиция ведет к повреждению легочных альвеол, сопровождающемуся интерстициальным отеком, увеличением альвеолокапиллярного барьера с последующим замещением эпителиоцитов гиперплазированными клетками. На 7-10 день могут появляться признаки фиброза. Респираторный тип гипоксии возникает в результате нарушения функции внешнего дыхания, то есть в результате нарушения вентиляции, диффузии, перфузии. Этиологическими факторами данного типа гипоксии являются следующие:
Основными патогенетическими механизмами данного типа гипоксии являются:
Циркуляторный тип гипоксии развивается при местных и общих нарушениях кровообращения. Выделяют ишемическую и застойную формы циркуляторной гипоксии. В случае, если нарушение гемодинамики развивается в сосудах большого круга кровообращения, насыщение крови кислородом может быть нормальным, однако, страдает его доставка к тканям. При нарушении кровообращения в сосудах малого круга страдает оксигенация крови. Этиологическими факторами циркуляторной гипоксии являются:
Циркуляторная гипоксия может возникнуть не только вследствие абсолютной, но и относительной недостаточности кровообращения, когда потребность тканей в кислороде превышает его доставку, примером может служить относительная недостаточность кровообращения в условиях стресса. Отличительной особенностью циркуляторной гипоксии является снижение массопереноса кислорода артериальной кровью и скорости его доставки к тканям. Содержание и напряжение кислорода при этой форме гипоксии может находиться в пределах нормооксических величин. Напряжение кислорода в венозной крови снижено. Вследствие этого венозно-альвеолярный и общий воздушно-венозный градиенты увеличены. При циркуляторной гипоксии выделяют:
Кровяной (гемический) тип гипоксии формируется при уменьшении кислородной емкости крови. Напомним, что кислородная емкость крови - количество О2, которое может быть связано с 100 мл крови. 1 г гемоглобина связывает 1,34 мл кислорода; 100 мл крови содержит около 15 г гемоглобина, следовательно, кислородная емкость крови около 20,1 мл кислорода. Гемическая гипоксия подразделяется на:
Этиологическими факторами анемической гемической гипоксии могут быть:
Гемическая (кровяная) гипоксия выступает результатом уменьшения кислородной емкости крови в результате: а) дефицита объема циркулирующих эритроцитов и низкой концентрации гемоглобина в крови; б) снижения кислородсвязывающих свойств гемоглобина. Кровяную гипоксию, которая развивается вследствие дефицита объема циркулирующих эритроцитов, называют анемической. Гемическая гипоксия развивается не только в результате блокады кислородсвязывающих свойств гемоглобина, но может быть следствием избыточного сродства гемоглобина к кислороду, снижающего восстановление гемоглобина и транспорт кислорода в клетку на периферии. Врожденные гемоглобинопатии, которые служат причиной гемической гипоксии подразделяют на два вида:
При гемоглобинопатиях, сопровождающихся цианозом, вследствие изменения аминокислотного состава молекулы гемоглобина в участке, прилегающим к гему (образование гемоглобина М), окисление железистой группы гемоглобина приводит к образованию метгемоглобина, чаще, чем при окислении нормального гемоглобина. Замещение в крови гемоглобина способного к переносу кислорода метгемоглобином приводит к падению кислородной емкости крови. Такие гемоглобинопатии наследуется по доминантному механизму. Побочные эффекты ряда лекарств и токсинов могут привести к патологическому росту содержания в крови метгемоглобина. К ним следует отнести в первую очередь нитриты и нитраты, анилиновые красители и сульфаниламиды. К образованию метгемоглобина в результате побочного действия лекарственных веществ и токсических эффектов химических соединений существует наследственная предрасположенность, за которую ответственен один из аллелей гетерозиготного гена. Известно более, чем 20 видов врожденных дефектов гемоглобина, при которых кривая диссоциации гемоглобина патологически сдвинута влево, что приводит к недостаточному высвобождению кислорода из соединения с гемоглобином на периферии и гипоксии. Угарный газ, окись углерода обладает сродством к гемоглобину в 240 раз большим сродства к нему кислорода. При парциальном давлении СО во вдыхаемой газовой смеси, составляющем 1/240 парциального давления в ней кислорода, окись углерода блокирует временное соединение кислорода с гемоглобином, образуя с ним относительно стойкое соединение, карбоксигемоглобин. В результате кровь содержит оксигемоглобин в количестве недостаточном для адекватного транспорта кислорода. Токсический эффект СО вызывает патологический сдвиг кривой диссоциации оксигемоглобина влево, что обостряет гипоксию вследствие недостаточного содержания оксигемоглобина в крови. Гипоксия вследствие инактивации гемоглобина развивается при острых и хронических интоксикациях. В качестве примера возникновения гемической гипоксии можно привести состояние возникающее при отравлении соединениями, содержащими NО2 группу. При остром и хроническом отравлении нитритами образуется метгемоглобин, в котором трехвалентное железо не обладает способностью присоединять кислород. Патогенез различных видов гемической гипоксии имеет общие признаки, заключающиеся в снижении кислородной емкости крови, вследствие чего ткани не получают достаточного количества кислорода, тканевое напряжение которого падает часто ниже критического уровня. Тканевая гипоксия. В зависимости от этиологии и патогенеза различают:
К первичной относят все те состояния, при которых происходит первичное повреждение аппарата клеточного дыхания либо на уровне клеточных органелл (митохондрий), либо на молекулярном уровне (ферментном уровне). Классическим примером тканевой гипоксии, при которой происходит инактивация дыхательных ферментов является отравление цианидами. Кроме того, первичная тканевая гипоксия может развиваться при отравлении лекарственными веществами, что важно знать врачам. Например, спирты, уретан и другие соединения блокируют дегидрогеназы. Ратион на уровне флавинадениндинуклеотида инактивирует дыхательную цепь. Тканевой тип гипоксии характеризует снижение способности клеток использовать кислород для биологического окисления. Кроме того, под тканевой гипоксией традиционно понимают системный гипоэргоз, обусловленный падением эффективности улавливания клеткой свободной энергии, высвобождаемой при биологическом окислении, что обычно связано с разобщением окисления и фосфорилирования. Использование клетками кислорода для биологического окисления нарушают: а) снижение активности энзимов цепи дыхательных ферментов митохондрий; б), сдвиги гомеостазиса внутриклеточной среды, блокирующие аэробное окисление; в) нарушения синтеза ферментов, участвующих в аэробном биологическом окислении и разрушение наружной клеточной и цитоплазматических, в том числе и митохондриальных, мембран. Следует заметить, что сдвига среды обитания клеток и внутриклеточной среды;- обусловленные гипоэргозом вследствие гипоксии, (лактатный метаболический ацидоз, рост содержания в клетке и в интерстиции аденозинмонофосфата и др.) сами по себе тормозят активность ферментов аэробного биологического окисления и растормаживают анаэробный гликолиз. Когда локальная гипоксия обусловлена ишемией, то из тканей и клеток в состоянии гипоксического гипоэргоза не удаляются с кровью как протоны, так и аденозинмонофосфат (АМФ). В результате концентрации в клетке свободных ионов водорода и АМФ растут в такой степени, что это тормозит уже последний источник свободной энергии анаэробный гликолиз. Нарушение способности клеток утилизировать кислород для биологического окисления и улавливать при нем свободную энергию в виде макроэргов, связанное с разрушением аппарата тканевого дыхания в результате гипоксии-ишемии, называют вторичной тканевой гипоксией. Во многом вторичная гипоксия связана со свободнорадикальным окислением наиболее в функциональном отношении активных фосфолипидов клеточных мембран, в том числе и митохондриальных. Свободнорадикальное окисление мембранных фосфолипидов предельно интенсивно после восстановления кровотока в ранее гипоксичных или ишемизированных тканях. При этом в тканях высока концентрация таких субстратов синтеза свободных кислородных радикалов как протоны, и есть второй его субстрат, кислород. В результате угнетения антиоксидантных систем гипоэргозом образование свободных кислородных радикалов при восстановлении притока артериальной крови после ишемии почти не ограничено, кроме свободнорадикального окисления фосфолипидов клеточных мембран к их деструкции может приводить широкий спектр патогенных воздействий, повреждающих клетку; гипо- и гипертермия, многочисленные экзогенные яды, проникающая радиация и т.д. Ингибирование ферментов биологического окисления как причина тканевой гипоксии происходит по трем основным путям. Первый состоит в специфическом связывании активных агентров дыхательных ферментов токсичными соединениями со вступать в стойкое соединение с активными центрами энзимов через специфическую реакцию, субстратами которой являются активный центр молекулы фермента и токсичный агент. Такими токсичными соединениями являются цианиды, содержащие токсичный ион циана, а также соединения, диссоциирующие с высвобождением токсичного сульфидного иона, некоторые антибиотики и др. Второй - это неспецифическое связывание функциональных групп белковой части молекулы фермента ионами тяжелых металлов и в результате реакции с алкилирующими агентами. Третий заключается в конкурентном торможении вследствие блокады активного центра ферментом «псевдосубстратом». «Псевдосубстрат» - это соединение, которое образуется на естественных путях метаболизма в норме или при их расстройствах, и, не являясь субстратом определенного энзима, блокирует его активный центр. Примером тут может служить ингибирование сукцинатдегидрогеназы малоновой и другими дикарбоновыми кислотами. Нарушения синтеза ферментов, участвующих в биологическом окислении, могут быть следствием качественного витаминного голодания, которое обуславливает недостаток в организме специфических нутриентов, необходимых для образования этих энзимов: витаминов группы В, тиамина, никотиновой кислоты и др. Алиментарная дистрофия как состояние системной недостаточности экспрессии генома клетки также нарушает синтез дыхательных ферментов. Снижение эффективности улавливания клеткой свободной энергии при аэробном биологическом окислении часто происходит через снижение сопряженности окисления и фосфорилирования на дыхательной цепи ферментов в митохондриях. При этом потребление кислорода растет, но аккумуляция свободной энергии в виде макроэргов падает, и ее значительная часть рассеивается в виде тепла. Разобщают аэробное биологическое окисление и фосфорилирование внутриклеточный ацидоз, избыток в клетке ионизированного кальция, неэстерифицированных жирных кислот, а также избыточное влияние на клетку адреналина и гормонов щитовидной железы. Таким свойством обладают и микробные токсины. Разобщение окисления и фосфорилирования может быть побочным эффектом лекарственных веществ (антибиотики и др.) При условии ненарушенных грубо легочного газообмена и транспорта газов с кровью напряжение кислорода в артериальной крови у больных с тканевой гипоксией находится в нормальных пределах при почти максимальном насыщении кислородом в ней гемоглобина. При этом растет напряжение О2 в смешанной венозной крови, и падает различие артериальной и смешанной венозной крови по содержанию в них кислорода. Выделяют четыре уровня адаптации к гипоксии (табл. 5.2 и рис. 5.1). Если доставка кислорода тканями и клетками становится неадекватной их потребностям вследствие снижения минутного объема кровообращения, концентрация гемоглобина в крови или расстройств легочного газообмена, то растет экстракция тканями кислорода из капиллярной крови. В результате снижается напряжение кислорода в смешанной венозной крови. При падении напряжения кислорода в смешанной венозной крови до 30 мм рт.ст. и ниже клетки начинают улавливать свободную энергию в ходе анаэробного биологического окисления, что приводит к аккумуляции в них и во внеклеточной жидкости молочной кислоты. При полном угнетении транспорта кислорода уровне всего организма при условии достаточного содержания в клетках глюкозы при анаэробном гликолизе образуются 3600 ммоль лактата в час. Внеклеточная жидкость содержит всего 375 ммоль бикарбонатного аниона, которое не могут связывать протоны, высвобождаемы при при диссоциации молочной кислоты. Это ведет к стремительному прогрессированию лактатного метаболического ацидоза, который сам по себе обусловливает цитолиз (гибель клеток) еще до того, как причинами гибели клеток становятся другие следствия гипоксии. При хронических циркуляторной и гемической гипоксии компенсаторный сдвиг вправо кривой диссоциации оксигемоглобина приводит к большему высвобождению кислорода на периферии при восстановлении оксигемоглоьина. Сдвиг кривой диссоциации оксигемоглобина враво вызывают: а) рост содержания в клетках 2,3-дифосфоглицерата; б) ацидоз. Таблица 5.2 Уровни адаптации к гипоксии
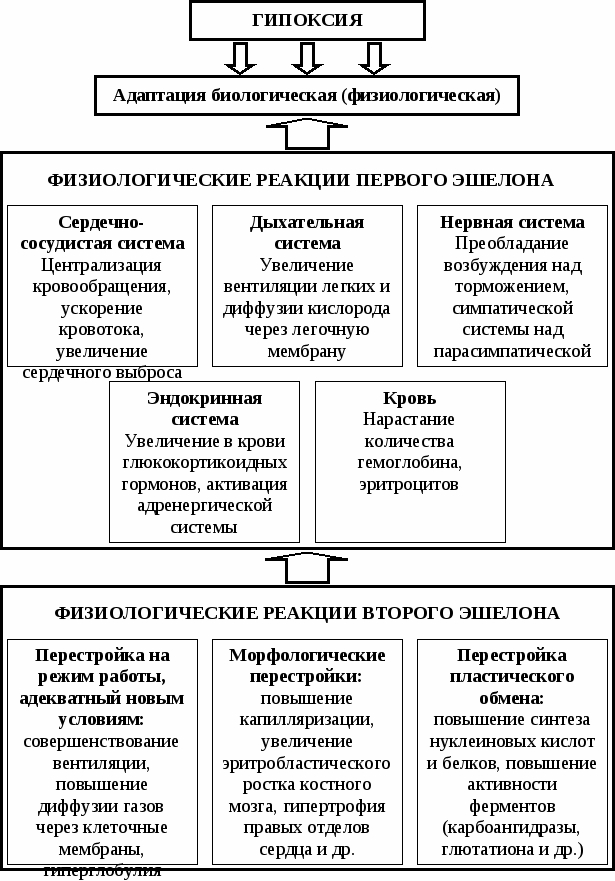 Рис. 5.1. Адаптация к гипоксии Достижение максимального уровня транспорта кислорода клеткам и тканям возможно при значениях гематокрита от 40 до 50%. При компенсаторном возрастании содержания в крови эритроцитов в ответ на хроническую гипоксию увеличение гематокрита до 55% приводит к росту транспорта кислорода от легких на периферию. Дальнейшее увеличение содержание эритроцитов и гематокрита приводит к падению минутного объема кровообращения из-за роста общего периферического сопротивления сосудов и расстройств микроциркуляции в результате увеличения вязкости крови. У больных частой причиной возникновения и обострения гипоксии служит усиление несоответствия между доставкой в клетки, ткани и резко возросшей в нем потребностью всего организма вследствие интенсификации аэробного обмена на системном уровне. Потребности организма в кислороде меняются в зависимости от потребности в кислороде на уровне всего организма. У больных острое повышение потребности в кислороде в частности вызывают судорожный синдром, мышечная дрожь и лихорадка. Особенно потребность в кислороде возрастает при диэнцефально-катаболическом синдроме у молодых больных после черепно-мозговых ранений и травм, при котором возникают одновременные лихорадка и судороги. Примером хронически повышенной потребности тканей в кислороде служит состояние аэробного обмена у больных с гипертиреозом. Наиболее ранний и эффективный механизм аварийной компенсации остро возросшей потребности клеток в кислороде – это возрастание минутного объема кровообращения. В условиях относительного покоя минутный объем кровообращения может возрастать в три-четыре раза, что в 3-4 раза повышает системный транспорт кислорода. При компенсации гипоксии, связанной с высокой потребностью в кислороде при физической нагрузке, именно рост минутного объема кровообращения служит детерминантой как максимального потребления кислорода легкими, так и максимального потребления кислорода на периферии. Гипервентиляция (рост минутного объема дыхания) также быстро развивается в ответ на развитие гипоксического состояния, как и ворастание минутного объема кровообращения. Если только у больного нет критических обструктивно-рестриктивных нарушений альвеолярной вентиляции, то снижение напряжения углекислого газа в артериальной крови выступает нормальным признаком срочной «аварийной» компенсации гипоксии. В этой связи следует возвращение значений напряжения углекислого газа в пределы средне-статистической «нормы» у больных с респираторной гипоксией признаком прогрессирования недостаточности внешнего дыхания, что у больных в астматическом статусе служит показанием для искусственной вентиляции легких. Напряжение кислорода в артериальной крови прямо пропорционально его поглощению легкими и обратно пропорционально потреблению кислорода организмом. Если интенсификация внешнего дыхания не приводит к росту поглощения кислорода легкими, которое соответствует возрастанию потребления кислорода организмом, то напряжение кислорода в артериальной крови падает. При сниженных функциональных резервах внешнего дыхания и кровообращения быстрое и значительное возрастание потребления кислорода организмом приводит к респираторной гипоксии. Одновременно с ростом потребления кислорода растет образование углекислого газа. При ограничении роста выделения углекислого газа вследствие обструктивно-рестриктивных расстройств альвеолярной вентиляции повышение его парциального давления в альвеолярной газовой смеси снижает в ней парциальное давление кислорода, и артериальная гипоксемия прогрессирует. При хронически и патологически высоком потреблении кислорода организмом (тиреотоксикоз, акромегалия) устойчивый компенсаторный сдвиг кривой диссоциации гемоглобина вправо обусловлен ростом содержания в эритроцитах 2,3-дифосфоглицерата (ДФГ). Этот неорганический фосфат образуется в эритроцитах при реализации там цикла биохимических реакций Раппорта-Люберинга. ДФГ связывается с бетта-цепью молекулы восстановленного гемоглобина, снижая его сродство к кислороду, что служит причиной большего восстановления гемоглобина на периферии с увеличением доставки кислорода непоредственно в клетку. Кроме того, ДФГ сдвигает кривую диссоциации оксигемоглобина враво через снижение рН в эритроцитах. Снижение синтеза дыхательных ферментов, вызывающее тканевую гипоксию наблюдается при авитаминозах. Особенно важен в отношении профилактики данного типа расстройств синтез рибофлавина и никотиновой кислоты. Вторичная тканевая гипоксия - состояние, при котором в результате несоответствия между скоростью доставки кислорода и потребностью в нем тканей напряжение кислорода падает. Для вторичной тканевой гипоксии характерными признаками являются:
На основании перечисленных признаков можно сделать заключение о том, что вторичная тканевая гипоксия может быть заключительным этапом любого из выше рассмотренных типов гипоксии. Существуют отличительные признаки первичной и вторичной тканевой гипоксии:
Общими признаками первичной и вторичной тканевой гипоксии являются:
Механизм снижения синтеза макроэргических соединений при вторичной тканевой гипоксии можно представить следующим образом. Синтез АТФ из АДФ и КФ и неорганического фосфора происходит благодаря тому, что окисленный цитохромС редуцируясь отдает свой электрон восстановленному НАДН, выделяемая при этом энергия идет на синтез АТФ. Редуцируемый цитохром С, соединяясь с кислородом переходит в окисленный с образованием двух молекул АТФ. Следовательно, данный процесс является кислородозависимым. При тканевом напряжении кислорода 30 мм рт. ст., что соответствует рО2 тканей человека в норме, цитохром С3 редуцирован на 30%, при рО2 12 и 6 мм рт. ст., цитохром С редуцирован на 33% и 40% соответственно. Параллельно с редукцией цитохрома наблюдается снижение фосфатного потенциала. Критический уровень потребления кислорода при вторичной тканевой гипоксии соответствует его напряжению в артериальной крови 50 мм рт. ст. Гипоксия нагрузки - состояние, возникающее при предъявлении повышенных требований к функциональной системе кислородного обеспечения организма. Усиление функции клеток обеспечивается повышенным расходованием энергии, утилизацией макроэргических соединений. Поставщиком АТФ является в норме окислительное фосфорилирование. Дыхание митохондрий зависит от фосфатного потенциала (АТФ/АДФхФн), степени редукции цитохрома С, наличия НАДН и НАД. В условиях гипоксии нагрузки, в начальный период усиления функций клеток, из-за инерционности системы дыхания 0 повышение скорости поэтапной доставки кислорода наблюдается не сразу, следовательно, в какой-то отрезок времени возникает несоответствие между скоростью потребления, которая значительно возрастает и скоростью доставки. В результате данного процесса запасы кислорода в клетке снижаются, что приводит к ограничению возможности потребления (за счет падения рО2). В дальнейшем данное несоответствие между скоростью доставки кислорода и его потреблением исчезает. Гипоксия при нагрузке малой интенсивности практически не отражается на общей скорости потребления кислорода, величина тканевого напряжения кислорода, поэтому такая форма гипоксии нагрузки может быть названа скрытой. Однако, в данном случае все же наблюдается снижение синтеза АТФ и фосфокреатина. При нагрузке средней интенсивности объем гипоксической зоны и степень рО2 в зоне наихудшего кровоснабжения зависят от количества функционирующих капилляров, скорости массопереноса кислорода. В данных условиях наблюдается вначале существенное увеличение скорости потребления кислорода. В случае значительного несоответствия между его доставкой и утилизацией по мере прогрессирования гипоксии, тканевое рО2 падает и снижается утилизация кислорода. При интенсивной физической нагрузке наблюдается вначале существенное увеличение скорости потребления кислорода. По мере прогрессирования гипоксии, утилизация кислорода снижается. Данное явление сопровождается значительным активированием гликолитического фосфорилирования, расходованием запасов макроэргических соединений. Патогенез различных видов гипоксии имеет общие признаки, заключающиеся в снижении кислородной емкости крови, вследствие чего ткани не получают достаточного количества кислорода, тканевое напряжение которого падает часто ниже критического уровня. Метаболические нарушения при гипоксии Чувствительность различных органов и тканей к гипоксии неодинакова и находится в зависимости от следующих факторов:
При гипоксии наиболее ранние изменения возникают в сфере энергетического обмена. При этом наблюдается дефицит макроэргических соединений: снижение запасов АТФ при одновременном увеличении АДФ, АМФ и Фн. Характерным признаком гипоксии является увеличение потенциала фосфорилирования: АДФ х Фн АТФ Ранним признаком является расходование фосфокреатина. В зависимости от тяжести гипоксии наблюдается снижение синтеза АТФ. Расходование запасов фосфокреатина сопровождается увеличением уровня активности креатинкиназы. В условиях дефицита кислорода происходит активирование безкислородных путей метаболизма. Наиболее распространенным видом анаэробного метаболизма в клетках тканей человека и животных является гликолиз (окисление глюкозы) и гликогенолиз (окисление гликогена). Напомним, что гликоген откладывается в цитозоле клеток в виде гранул диаметром 100-400 А в форме β-частиц. Имеются также β-частицы диаметром до 1000 А, которые наиболее часто встречаются в цитозоле гепатоцитов человека. Кроме того, имеются гликоген-мембранные комплексы, например, в сердечной мышце. Каждая из перечисленных форм запасов гликогена определяет метаболические пути его утилизации при гипоксии. Модификация гликолитического пути заканчивается сходством их заключительного этапа. У человека этот процесс оканчивается образованием лактата, что способствует снижению цитоплазматического рН, развитием ацидоза. Катаболизм липидов в клетке начинается с экзогенных свободных жирных кислот или эндогенных триацилглицеролов. У млекопитающих большинство тканей (кроме мозга) способно к мобилизации триацилглицеролов. Главным депо липидов служит жировая ткань, которая способна удовлетворить всю потребность организма в жирных кислотах. Жирные кислоты окисляются до ацетил-СоА, затем происходит перенос ацильных групп в митохондрию, где происходит β-окисление. В условиях дефицита кислорода происходит накопление недоокисленных продуктов обмена, таких как β-оксимасляная кислота, ацетоуксусной кислоты. Дефицит кислорода приводит к нарушению обмена белков. Организм может катаболизировать белки и аминокислоты, которые поступают из двух источников:
В организме происходит гидролиз белка до аминокислот, последние расщепляются до аммиака. Аммиак может быть использован в организме для синтеза азотсодержащих соединений, большая часть его выделяется в виде азотистых продуктов. У большинства позвоночных аммиак превращается в мочевину. На синтез одной молекулы мочевины расходуется 4 молекулы АТФ. В условиях дефицита энергии происходит нарушение утилизации аммиака. Следовательно, в условиях гипоксии будет наблюдаться увеличение концентрации аммиака в тканях и крови, нарастание катаболических процессов над анаболическими. В условиях гипоксии наблюдаются нарушения электролитного обмена. Основными причинами, вызывающими данное явление служат:
В условиях гипоксии наблюдается увеличение свободно-радикального окисления. В основе данного процесса лежат следующие механизмы:
В условиях гипоксии наблюдается снижение активности антиоксидантных систем клетки, в частности уменьшение уровня активности супероксиддисмутазы и глютатионпероксидазы. Активирование свободно-радикального окисления приводит к следующим явлениям:
Нарушение клеточных процессов при гипоксии При гипоксии наблюдаются нарушения функционирования цитоплазматических и эндоплазматических мембран, которые могут проявляться в виде нарушения их проницаемости, снижения величины потенциала покоя клеточной мембраны, изменения функционирования транспортных систем. В митохондриях наблюдается нарушение окислительного фосфорилирования со снижением степени сопряженности данного процесса, изменение проницаемости мембран митохондрий. Морфологически гипоксические изменения в митохондриях выявляются в виде их набухания, изменения электронномикроскопической структуры мембран. При тяжелой гипоксии, сопровождающейся значительным падением тканевого напряжения кислорода (ниже критического уровня) происходит значительное уменьшение количества крист митохондрий, ─ возможно снижение количества митохондрий. При гипоксии наблюдаются изменения в саркоплазматическом ретикулуме, в результате чего происходит нарушение микросомального окисления, нарушение транспортных процессов в клетке, изменение обмена ионов внутри клетки (между ее компартментами). Нарушение функций лизосом сопровождается лабилизацией лизосомальных ферментов с увеличением уровня их активности в цитоплазме. В результате данных процессов могут наступать необратимые нарушения функций клеток, их повреждение. Гипоксия приводит также к нарушению функций клеточного ядра, основным следствием данного процесса может быть изменение специфических функций клеток. Морфологически гипоксические изменения в клетке выявляются в виде 3 гиперхроматоза ядра, возможен пикноз ядра. Тяжелая гипоксия может приводить к деструкции клеточного ядра. Перечисляя изменения в клетке при гипоксии следует отметить, что общим универсальным механизмом гипоксического повреждения клеток является увеличение пассивной проницаемости цитомембран. Описывая изменения в клетках при гипоксии следует отметить, что наиболее чувствительны к гипоксии клетки нейроциты, особенно нейроны коры головного мозга, затем нейроны ствола и спинного мозга. Наименее чувствительны к гипоксии костные образования, хрящи, сухожилия, мышцы, кожа. Принципы предупреждения и коррекции гипоксических состояний Одним из основных способов адаптации организма к гипоксии является уменьшение функциональной активности органов и систем, переход их на экономное расходование кислорода и субстратов биологического окисления. Основные принципы профилактики гипоксии включают:
Основные принципы терапии гипоксии включают:
|