учебник по патфизу. Учебник для слушателей и курсантов военномедицинской академии и военномедицинских институтов под редакцией проф. В. Ю. Шанина
 Скачать 4.96 Mb. Скачать 4.96 Mb.
|

|
Глава 10. РАССТРОЙСТВА КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ Основные механизмы удержания концентрации протонов во внеклеточном секторе и клетках в нормальных пределах. Параметры кислотно-основного состояния и виды его расстройств Концентрацию протонов во внеклеточной жидкости ([Н+]) функциональные системы удерживают на уровне 40 нмоль/л, который является низшим относительно высвобождения свободных ионов водорода в клетки и во внутреннюю среду (7х107 нмоль каждый день). При обычном питании высвобождение протонов составляет 1 ммоль/кг массы тела в день, что в 106 раз и превышает общее содержание протонов во внеклеточной жидкости. Свободные ионы водорода связываются буферными системами: 40% всех высвобождаемых протонов нейтрализует главная внеклеточная буферная система угольной кислоты и гидрокарбоната натрия, а 60 % - внутриклеточные буферные системы. После взаимодействия протона с буферной системой он переходит во временное состояние связывания и нейтрализации. Буферные системы - это первая защитная линия в противодействии тенденции роста концентраций протонов в клетках и во внутренней среде, который является фактором апоптоза и цитолиза, доя поддержания нормального кислотно-основного состояния необходимо выведение протонов из форм их временного связывания во внешнюю среду. Легкие - это эффектор функциональной системы удержания концентрации протонов во внутренней среде в нормальных пределах. Через легкие происходит выведение углекислого газа, которое обусловливает постоянный сдвиг реакций системы бикарбонатного буфера вправо: H+ + HCO3- = H2O + CO2 Почки восполняют потери бикарбонатного аниона, используя глутамин как субстрат образования аммиака и аммониевого катиона (NH4+). Глутамин образуется в печени. В физиологических условиях печень - это главный локус высвобождения протонов. При образовании аммониевого катиона, в составе которого протоны выделяются вместе с мочой, в почках образуется один бикарбонатный анион. Для удержания [Н+] в нормальных пределах образование бикарбонатных анионов должно соответствовать их тратам для временной нейтрализации протонов. Кислотно-основное состояние больного характеризуют величины трех его параметров:
Функциональную связь между ними отражает уравнение Гендерсона-Гассельбаха: [Н+](нмоль/л)=23,9 - РаСО2(мм рт. ст.)/[НСО3-](ммоль/л). В желудочное содержимое каждые сутки попадает 150 ммоль протонов. Выведение одного протона в просвет желудка обкладочной клеткой сопровождается образованием одного гидрокарбонатного аниона, который поступает во внеклеточную жидкость. Тонкая кишка и поджелудочная железа секретируют бикарбонатные анионы, нейтрализуя протоны в просвете кишечника. В физиологических условиях те бикарбонатные анионы, которые не связывают протоны, поступившие в кишечник из желудка, реабсорбируются в кровь. Потери кислого содержимого желудка усиливают образование бикарбонатного аниона в париетальных клетках. Это повышает [HCО3-]. Потери содержимого кишечника (диарея, кишечные свищи и др.) уменьшают реабсорбцию бикарбонатного аниона до просвета желудочно-кишечного канала. Это снижает [НСО3-]. Микроорганизмы в просвете кишечника могут образовывать не усваиваемые организмом органические кислоты, что служит причиной метаболического ацидоза. Полное окисление органических анионов из состава пищи микроорганизмами в просвете кишечника до углекислого газа и воды ведет к аккумуляции в нем бикарбонатного аниона, что вызывает метаболический алкалоз. Для оценки кислотно-основного состояния необходимо определение четырех его параметров (табл. 10.1). Таблица 10.1 Нормальные величины параметров кислотно-основного состояния
Выделяют четыре вида нарушений кислотно-основного состояния: метаболический ацидоз, метаболический алкалоз, респираторный ацидоз, респираторный алкалоз.
Патологические сдвиги концетрации протонов во внеклеточной жидкости и плазме крови вызывают реакций организма, которые направлены на возвращение [Н+] в нормальные пределы, что достижимо только при хроническом респираторном алкалозе. Метаболический ацидоз Метаболический ацидоз - это патологическое состояние, которое характеризуют рост концентрации протонов ([Н+])в клетках и внеклеточной жидкости, а также снижение в них содержания бикарбонатного аниона ([НСО3-]). К метаболическому ацидозу приводят задержка в организме сильно диссоциирующих кислот и (или) потеря ими бикарбонатного аниона. Для определения основного звена патогенеза метаболического ацидоза необходимо определить анионный пробел плазмы, Число анионов всегда равно числу катионов как в клетках, так и во внеклеточной жидкости. Если из величины содержания во внеклеточной жидкости и жидкой части плазмы их главного одновалентного катиона ([Na+]) вычесть общее содержание в них главных одновалентных анионов, хлоридного ([C1]) и бикарбонатного ([НСO3]), то мы получим значение анионного пробела плазмы (АПП): AПП = [Na+] - ([Cl-] + [HCO3-]). Во внеклеточной жидкости могут диссоциировать эндо- или экзогенные кислоты. Диссоциация кислот повышает во внеклеточной жидкости количество протонов и анионов. При этом содержание катиона натрия, во внеклеточной жидкости остается на прежнем уровне, так как механизмы активного переноса ионов через наружную клеточную мембрану удерживают натрий во внеклеточном секторе. В результате, несмотря на то, что общее содержащая анионов во внеклеточной жидкости остается неизменным, АПП становится на 1 ммоль больше (рис. 10.1), Поэтому рост АПП выше верхнего Предела нормальных колебаний (10-14 ммоль/л) свидетельствует о повышенном высвобождении во внутреннею среду или поступлении из внешней диссоциирующих кислот, которые вызывают метаболический ацидоз. Если АПП не растет, то причина ацидоза - этопотеря организмом бикарбонатного аниона. Содержание альбуминовых анионов во внеклеточной жидкости и плазме при нормальной концентрации белков в крови составляет 12ммоль/л. Гипоальбуминемия уменьшает АПП без нарушений кислотно-основного состояния. Поэтому у больных с гипоальбуминемией диссоциация органических кислот в клетках и во внеклеточной жидкости вызывает метаболический ацидоз при АПП в “нормальных” пределах. 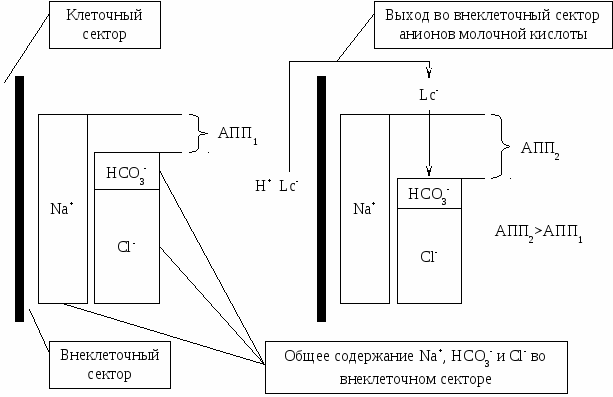 Lс- - анионы молочной кислоты Рис. 10.1. Рост анионного пробела плазмы (АПП) при лактатном ацидозе Этиопатогенетическая классификация метаболического ацидоза выделяет два его вида: нетаболический ацидоз с ростом АПП и метаболический ацидоз с нормальным АПП. Наиболее частая причина лактатного метаболического ацидоза - это недостаточный транспорт в клетку кислорода. Дефицит транспорта кислорода определяют артериальная гипоксемия, а также нарушения системного и периферического кровообращения. Накопление лактата в цитозоле клеток происходит только при замедлении трансформации лактата в пируват, которая невозможна без сопряженного с ней окисления восстановленной формы никотинамидадениндинуклеотида (НАДН). Гипоксия через падение напряжения кислорода в митохондриях ведет к накоплению в них НАДН, блокируя передачу протона от восстановленной формы НАДН вцитозоле к ее окисленной форме в митохондриях. Это повышает содержание НАДН в цитозоле и ведет к накоплению в клетке молочной кислоты. Концентрация молочной кислоты в клетке растет вследствие угнетения превращения лактата в пируват, обусловленного yгнетением или прекращением окисления НАДН. Второй непосредственной причиной роста содержания лактата в цитозоле клеток может быть аккумуляция в них пировиноградной кислоты. Лактатный ацидоз, связанный с гипоксией, - это лактатный ацидоз типа А. Острая респираторно-циркуляторная гипоксия как причина лактатного ацидоза может вызывать высвобождение во внутреннюю среду протонов со скоростью 60 ммоль/мин, чему не в состоянии противостоять система бикарбонатного буфера внеклеточной жидкости. Острый лактатный ацидоз можно считать одним из основных механизмов необратимости тяжелого шока и клинической смерти. Локальная ишемия не приводит к лактатному ацидозу типа А, так как одновременно с прекращением доставки в клетки кислорода в них перестает поступать глюкоза как предшественник молочной кислоты на пути ее образования. Кроме того печень трансформирует молочную кислоту, высвобождаемую локальным очагом ишемии, в глюкозу. Лактатный ацидоз типа В - это следствие повышенного высвобождения молочной кислоты печенью. Его может вызвать угнетшие трансформации лактата в глюкозу гепатоцитами на уровне всей печени при печеночной недостаточности. Лактатный ацидоз может быть следствием недостатка в организме тиамина, вызванного витаминным голоданием или алкоголизмом. Дефицит тиамина приводит к лактатному ацидозу через угнетение утилизации пирувата на путяхобмена веществ. Аналогичным образом вызывают лактатный ацидоз типа Б наследственные нарушения обмена веществ: непереносимость фруктозы и недостаточная активность некоторых ферментов, участвующих в глюконеогенезе. Отравление этанолом черезего интенсивную переработку в печени ведет к накоплению в цитозоле гепатоцитов НАДН, что вызывает накопление лактата. Опухоли печени при определенной распространенности поражения ее паренхимы уменьшают очищение плазмы крови от лактата печенью. Падение клиренса лактата печенью приводит к лактатному ацидозу (типа Б). Опухоли вызывают лактатный ацидоз типа Б, высвобождая метаболиты, которые угнетают глюконеогенез в печени. 1К ним, в частности, относят метаболит триптофана, угнетающий активность ключевого фермента глюконеогенеза - фосфоенолпируваткарбоксикиназы. Злокачественные опухоли большой массы могут интенсивно высвобождать молочную кислоту. Высвобождение лактата опухолями может преобладать над способностью печени очищать кровь от лактата ивызывает метаболический адидоз. Некоторые избактерий в просвете желудочно-кишечного тракта могут трансформировать клетчатку, которую содержит пища, в органические кислоты, Образование органических кислот бактериями из клетчатки становится особенно интенсивным при замедлении прохождения пищевых масс по желудочно-кишечному каналу (слепые петли кишки, обструкция, непроходимость) и изменении состава кишечной флоры под влиянием терапии антибиотиками. В основном бактерий образуют из клетчатки D-изомер молочной кислоты. Так как организм человека метаболизирует этот изомер лактата медленнее, чем эндогенный L-изомер, то абсорбция D-изомера вызывает метаболический ацидоз с увеличенным АПП. Существующие способы опре-деленйя концентрации лактата во внеклеточной жидкости и жидкой части плазмы крови ориентированы на L-изомер, что затрудняет выявление метаболического ацидоза, вызванного дисбактериозом в просвете кишечника. Часть D-изомера лактата выводится из крови почками. Этo создает несоответствие между увеличением АПП и снижением концентрации бикарбонатного аниона в плазме крови. Выделяют два вида кетоацидоза:
К кетоацидозу первого вида приводит гипогликемия как причина угнетения секреций инсулина. Голодание редко снижает содержание глюкозы в плазме крови до уровни более низкого, чем 3 ммоль/л. При метаболическом кетоацидозе как следствии голодания концентрация бикарбонатного аниона в плазме не падает ниже 18 ммоль/л, а ее анионный пробел не становится большим, чем 19 ммоль/л. При таком умеренно выраженном метаболическом ацидозе внутривенное вливание раствора глюкозы прекращает кетогенез и устраняет метаболический ацидоз. Если гипогликемия как причина кетоацидоза связана с нарушениями накопления и хранения гликогена в гепатоцитах, то она часто сочетается с низким уровнем глюконеогенеза в печени. При этом снижено образование глюкозы печенью из лактата. В результате метаболический ацидоз обусловлен как кетогенезом, так и диссоциацией молочной кислоты во внеклеточной жидкости (лактатный метаболический ацидоз типа Б). Поэтому при таком патогенезе метаболического ацидоза, который вызывает действие двух механизмов, в отличие от кетоацидоза, вызванного голоданием [НСО3-] часто ниже, чем 18ммоль/л. Этиопатогенетическая терапия основного заболевания и внутривенное вливание растворов глюкозы в частности устраняют кетоацидоз данного генеза. Этанол как непрямой адреномиметик тормозит секрецию инсулина через возбуждение альфа-адренорецепторов бета-клеток поджелудочной железы, Таким же свойством обладают адреномиметики. Падение секреции инсулина усиливает секрецию гл юкагона, который увеличивает кетогенез. Кроме того, адренергическое торможение секреции инсулина может быть следствием стрессорной реакции в ответ на дефициты внеклеточной жидкости и объема циркулирующей крови. Поэтому, если при отравлении этанолом частая и обильная рвота ведет к дефициту объема внеклеточной жидкости, то кетоацидоз вследствие отравления этиловым спиртом развивается быстрее. При такой этиологии кетоацидоза он может быть тяжелым метаболическим ацидозом. У таких больных часто выявляют гипергликемию вследствие усиления гликогенолиза и глюконеогенеза в результате падения секреции инсулина. Внутривенное вливание таким пациентам раствора глюкозы устраняет кетоацидоз только тогда, когда его причина - гипогликемия. Интенсивная терапия при кетоацидозе, вызванном отравлением этанолом, в основном сводится к вдутривенному вливанию растворов натрия и калия хлоридов, направленному на устранение дефицитов объема внеклеточной жидкости и калия в организме. Почечная недостаточность может приводить к метаболическому ацидозу с увеличением АПП. У здоровых людей и у пациентов с хронической почечной недостаточностью высвобождение протонов печенью при утилизации аминокислот, поступивших в организм с пищей, составляет около 1 ммоль/кг массы тела за сутки. Это количество протонов нейтрализует эквивалентное число гидрокарбонатных анионов. Потери гидрокарбонатного аниона из внеклеточной жидкости восполняет образование новых анионов почками, сопряженное с образованием и экскрецией NН4+. По мере прогрессирования хронической почечной недостаточности падают экскреция NH4+ и образование почками бикарбонатных анионов. Снижение концентрации бикарбонатного аниона во внеклеточной жидкости и жидкой части плазмы ведет к метаболическому ацидозу. Одновременное снижение скорости клубочковой фильтрации служит причиной падения экскреции органических анионов, что повышает АПП. При этом рост АПП и снижение концентрации бикарбонатного аниона вo внеклеточной жидкости относительно независимы друг от друга и не находятся в прямой связи. Почечный канальцевый ацидоз Почечный канальцевый ацидоз - это метаболический ацидоз вследствие падения экскреции протонов с мочой без падения скорости клубочковой фил ьтрации.Снижение экскреции протонов с мочой приводит к снижению образования в почках биканатного аниона. В результате падает [НСО3-], и возникает метаболический ацидоз без роста АПП. Нормальная скорость клубочковой фильтрации при почечном канальцевом ацидозе обеспечивает обычное для физиологических условий выведение с мочой фосфатного и сульфатного анионов. Данные анионы не задерживаются в организме, и АПП не растет. Для поддержания электронейтральности внеклеточной жидкскти, несмотря на низкий уровень [НСО3-], растет реабсорбция хлоридного аниона из просвета канальцев нефрона. Во внеклеточной жидкости увеличивается содержание хлоридных анионов, которые возмещают общий недостаток анионов, возикший из-за низкого образования НСО3-в почках. Поэтому почечный канальцевый ацидоз характеризует гиперхлоремия. Выделяют четыре типа почечного канальцевого ацидоза(I,II,III,IV). Почечный канальцевый ацидоз I (ПКА I) обычно наследуется по аутосомно-доминантномутипу. Причина высокого рН при ПКА I – этоизбыточная обратная диффузия протонов из просвета собирательных канальцев в кровь. Кроме того, причиной канальцевого ацидоза I может быть недостаточность активного трансмембранного переноса протонов в просвет канальцев против значительного градиента их концентраций. В основном выделение протонов происходит в собирательных канальцах, где протоны активно секретируются и выводятся с мочой в составе аммониевого катиона и титруемых кислот. Аммиак вместе с протонами служат субстратами для образования временно существующего аммониевого катиона, выводимого во внешнюю среду в составе мочи. Все эти физиологические процессы необходимым услоием имеют нормальный и достаточно низкий рН мочи. Рост рН мочи у больных с ПКА I угнетает данные процессы экскреции протонов. Экскреция аммониевого катиона угнетается в меньшей степени и может даже расти от физиологического уровня для компенсации метаболического ацидоза. При метаболическом ацидозе и патологически высоком рН мочи у больных с ПКА I падает реабсорбция кальция из канальцев нефрона. В результате возникает гтперкальциурия. В физиологических условиях 40 % свободных ионов кальция в канальцах нефрона связываются цитратными анионами. При ПКА I из-за гиперкальциурии свободный кальций в большей степени связывается фосфатным анионом. Это ведет к большему образованию в моче кальция фосфата и кальци-нозу почечных сосочков. Гиперкальциурия вызывает гипокальцие-мию и вторичный гиперпаратиреоз. Нарушения обмена кальция ведут к задержке роста у детей и остеомаляции у взрослых. Причиной задержки роста и остеомаляции при ПКА I может быть снижение синтеза (витамин D)-гормона. ПКА I характеризуют взаимосвязанные патологические рост рН мочи, падение водного диуреза, снижение осмоляльности мочи, полиурия и гипокалиемия (следствие избыточной ренальной экскреции калия). Гипокалиемия и ацидоз при возникновении сопутствующих заболеваний могут стать звеньями танатогенеза. Иногда ПКА I может быть следствием семейной наследственной идиопатической гиперкальциурии. Кроме того, ПКА I вызывают такие наследственные заболевания, как эллиптоцитоз, болезнь Фабри, синдром Элерса-Данлоса-Русакова5, галактоземия, медуллярная губчатая почка. Почечный канальцевый ацидоз второго типа (ПКА II) обычно представляет собой одно из следствий общих расстройств функциональной выделительной системы на уровне проксимальных канальцев нефрона (синдром Фанкони и др.). В результате данных расстройств в составе конечной мочи патологически растет концентрация аминокислот, фосфатного аниона и мочевой кислоты. Возникая у новорожденных, ПКА II в дальнейшем по ходу онтогенеза может подвергнуться обратному развитию. Ведущим звеном патогенеза ПКА II является недостаточная реабсорбция бикарбонатного аниона в проксимальных сегментах нефрона при нормальном содержании НСО3- в плазме крови. В результате возникает не существующая в физиологических условиях бикарбонатурия, которая исчезает в ответ на снижение концентрации бикарбонатного аниона в плазме крови. Бикарбонатурия может привести к гипокалиемии, так как бикарбонатные анионы увлекают за собой в состав конечной мочи катионы калия. При ПКА II гиперкальциурия минимальна, и мочевые камни не образуются. Почечный канальцевый ацидоз IV типа (ПКА IV) характеризует гиперкалиемия без роста АПП. От почечного канальцевого ацидоза типа I данный вид канальцевого ацидоза отличает рост содержания протонов в моче вответ на усиление метаболического ацидоза. В отличие от ПКА II при канальцевом ацидозе четвертого типа не бывает значительной бикарбонатурии. При этомПКА IV можно считать состоянием, одним из звеньев патогенеза которого является низкое образование аммиака в почках. Гиперкалиемия при ПКА IV служит фактром метаболического ацидоза, угнетая обращение аммиака, а значит и выведение протонов с мочой в составе аммониевого катиона. У большинстш больных с ПКА IV в плазме крови выделяют низкие активность ренина и концентрацию алдостерона (гипонатриемический гипоальдостеронизм). Активность ренина и конценрация гормона не растут даже вответ на дефщит в организме натрия и объема внеклеточной жидкости. В данном случае заместительная терапия препаратами со свойствами минералокортикоидов устраняет метаболический ацидоз. У меньшей части больных ПКА IV связан с низкой реакцией тубулярныхэпиителиоцитов на действие альдостерона (минералокортикоид-резистивная гиперкалиемия). У таких больных препараты со свойствами минералокортикоид не устраняют ПКА. В частности, ПКА IV вследствие минералокортикоид-резистивной гиперкалиемии развивается после патологических изменений дистальных сегментов нефрона в результате какого-либо первичного заболевания почек. У таких больных патологически измененные почки теряют натрий, а в ответ на дефицит натрия и объема внеклеточной жидкости в плазме крови растут активность ренина иконцентрация альдостерона. У части больных с ПКА IV и минералокортикоидрезистивной гиперкалиемией нет потерь натрия, и метаболический ацидоз выражен умеренно. Патогенез ПКА IV такого вида предположительно связан с патологически высокой проницаемостью стенок дистальных сегментов нефрона для хлоридного аниона (рис. 10.2). ПКА III - это ПКА I у детей. 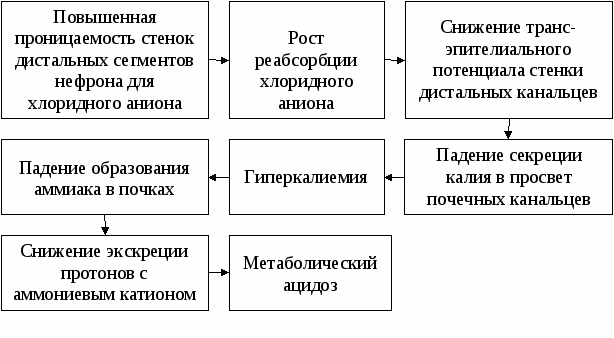 Рис. 10.2. Патогенез (предположительный) ПКА IV без дефицитов натрия, объема ццркудирующей крови и дераичадйс по отношению к ПКА заболеваний почек Метаболический алкалоз Метаболический алкалоз вызывают увеличение содержания во внеклеточной жидкости и жидкой части плазмы крови бикарбонатных анионов и его следствие - одновременное падение в них концентрации протонов. Рост [НСО3-] обусловливают:
Необходимое условие сохранения общего содержания бикарбонатного аниона в организме на неизменном уровне - это ненарушенное образование бикарбонатных анионов почками. Клинико-патофизиологическая классификация метаболического алкалоза выделяет два его вида.
Метаболический алкалоз первого вида чаще является следствием потерь желудочного содержимого вследствие обильной рвоты или по гастральному зонду (рис. 10. 3). Потери содержимого желудка вызывают дефицит объема внеклеточной жидкости. Потери хлоридного аниона с содержимым желудка снижают концентрацию Сl- до уровня более низкого, чем 20 ммоль/л. Причиной метаболического алкалоза первого вида может быть побочное действие диуретиков. 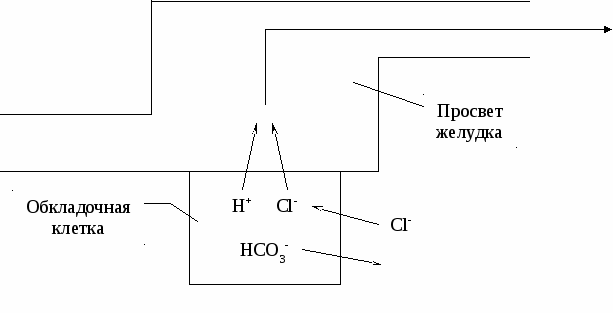 Рис. 10.3. Потери содержимого желудка как причина метаболического алкалоза и гипохлоремии Метаболический алкалоз второго вида встречается реже. Его обусловливают:
Потеря с содержимым желудка одного протона ведет к высвобождению одного бикарбонатного аниона в париетальных клетках желудрчного эпителия. Для поддержания электронейтральности внутри эпителиоцита бикарбонатный анион уходит во внеклеточную жидкость, что повышает [НСО3-]. Для поддержания электронейтральности внеклеточной жидкости “в обмен” на гидрокарбонатный анион в просвет желудка уходит анион Cl-. Это снижает содержание Cl- во внеклеточной жидкости и плазме крови (гипохлоремический алкалоз) (рис 6). Потери содержимого желудка при частой рвоте и по гастральному зонду снижают содержание во внеклеточной жидкости хлоридного аниона, который в ней замещается бикарбонатным анионом. Если при этом не падают объем внеклеточной жидкости и скорость клубочковой фильтрации, то рост [НСО3-] повышает содержание бикарбонатного аниона в ультрафильтрате почечных клубочков. При этом секреция протонов в просвет канальцев проксимальных сегментов нефрона не возрастает, так как еще не действуют ее стимулы - дефицит объема внеклеточной жидкости и снижение содержания в ней калия. В результате низкой секреции протонов относительно содержания НСО3- в ультрафильтрате в проксимальных сегментах нефрона часть бикарбонатных анионов остается в просвете канальцев и увлекает за собой в состав конечной мочи катионы натрия. Поэтому у больных с потерями содержимого желудка до появления признаков дефицита объема внеклеточной жидкости и гиповолемии фиксируют высокое содержание натрия в конечной моче. Тяжелый метаболический алкалоз первого вида характеризуют гипокалиемия и признаки дефицита объема внеклеточной жидкости. По мере увеличения потерь содержимого желудка растут [НСО3-] и выделение натрия с мочой. В организме снижается содержание натрия. Дефицит натрия вызывает дефицит объема внеклеточной жидкости и активацию секреции альдостерона. Усиление действия альдостерона на эпителиоциты почечных канальцев повышает выведение калия cмочой. В результате возникает гипокалиемия. Гипокалиемия усиливает метаболический алкалоз через усиление секреции протонов в просвет канальцев. Возрастающие потери натрия с желудочным содержимым постепенно приводят к значительному уменьшению объема внеклеточной жидкости, который ограничивает выделение бикарбонатного аниона с мочой. Во-первых, дефицит объема внеклеточной жидкости уменьшает скорость клубочковой фильтрации и число би-карбонатных анионов в составе ультрафильтрата. Во-вторых, дефицит объема внеклеточной жидкости усиливает секрецию минералокортикоидов. Повышенная концентрация минералокортикоидов в циркулирующей крови увеличивает секрецию протонов в собирательных канальцах, что усиливает реабсорбцию бикарбонатного аниона и экскрецию калия. Гипокалиемия как результат побочного действия диуретиков (в том числе и в результате стимуляции дефицитом объема внеклеточной жидкости секреции альдостерона) усиливает метаболический алкалоз. Мочегонные средства вызывают метаболический алкалоз в основном через снижение объема внеклеточной жидкости. Дело в том, что диуретики приводят к потере катиона натрия и хлоридного аниона, не увеличивая экскреции НСО3-. Потери натрия снижают объем внеклеточной жидкости при неизменном числе в ней бикарбонатных анионов. В результате растет [НСО3-] и развивается метаболический алкалоз. Кроме того, дефицит объема внеклеточной жидкости через снижение объема плазмы крови уменьшает скорость клубочковой фильтрации. Падение скорости клубочковой фильтрации обусловливает неэффективность защитной реакции увеличения выделения НСО3- с мочой в ответ на рост уровня [НСО3-]. Повышенный диурез как побочный эффект диуретиков определяет дефициты объемов внеклеточной жидкости и плазмы крови. Это служит причиной усиленной секреции минералокортиковдов. Рост содержания минералокортикоидов в крови увеличивает секрецию протонов в просвет канальцев нефрона. Это повышает концентрацию во внутренней среде бикарбонатных анионов, образованных почками. Если концентрации в моче Na+ и Cl- определяют во время продолжающегося действия диуретиков, то они больше, чем 20 ммоль/л. В другой момент они ниже этого уровня, как и при гипохлоремическом алкалозе, вследствие потерь содержимого желудка. У некоторых больных внеклеточная жидкость может содержать экзогенные анионы (в результате приема или введения антибиотиков и т. д.), которые попадают в состав ультрафильтрата, но не реабсорбируются из просвета канальцев. Эти анионы, находясь в просвете канальцев, препятствуют ребсорбции натрия в проксимальных сегментах нефрона. Растет число катионов натрия, поступающих в дистальные сегменты нефрона. Это повышает реабсорбцию натрия, образование НСО3-, а также секрецию в просвет канальцев протонов и калия. Рост реабсорбции натрия в дистальных сегментах нефрона не предотвращает потерь катиона с мочой. Это снижает объем внеклеточной жидкости, и его дефицит обостряет метаболический алкалоз. При хронической дыхательной недостаточности задержка во внутренней среде 6икарбонатного аниона почками конденсирует респираторный ацидоз. При этом бикарбонатный анион замешается в моче хлоридным анионом. Представим, что у пациента после устранения гипокапнии есть дефицит объема внеклеточной жидкости. Мы уже знаем, чтодефицит объема этой жидкости caм по себе служит причиной метаболического алкалоза. Кроме того, в данной ситуации он препятствует выведению из внеклеточной жидкости с мочой бикарбонатных анионов, которые были задержаны во внутренней среде во время гиперкапнии. Все это приводит ic постгиперкапническому метаболическому алкалозу. Следует заметить, чтодлительной гиперкапнии часто сопутствуют критические состояния, вызывающие обезвоживание и дефицит объема внеклеточной жидкости. Метаболический алкалоз, который не устраняет внутривенная инфузия изоосмодялыдах относительно внеклеточной жидкости растворов, характеризуют:
Необходимое условие метаболического алкалоза второго вида при любой его этиологии - это поступление катиона натрия и хлоридного аниона в собирательные канальцы коркового слоя при действии на эти сегменты нефрона, циркулирующих с кровью минералокортикоидов в повышенной концентрации. Это приводит к увеличению реабсорбций натрия и усилению экскреции протонов и калия. В результате роста выделения калия развивается гипокалиемия. Гйпокалиемия как стимул образования аммиака в эпителиоцитах канальцев увеличивает генерацию бикарбонатных анионов почками. Увеличение образования аммиака ведет к росту выделения протонов в виде аммониевых катионов и метаболическому алкалозу. Одновременно гипокалиемия снижает скорость клубочковой фильтрации, что уменьшает эффективность бикарбонатурии как компенсаторной реакции на метаболический алкалоз. Рост активности минералокортикоидов в циркулирующей крови обусловливают:
При реноваскулярной артериальной гипертензии и ренинпродуцирующих опухолях почек рост концентрации альдостерона в крови обусловлен высокой активностью в ней ренина. Ятрогенный метаболический алкалоз второго вида вызывает действие лекарств, обладающих свойствами минералокортикоидов. До сих пор неясным остается патогенез метаболического алкалоза второго вида, связанного с дефицитом в организме магния. При синдроме Бартера, этиология и патогенез которого также остаются неясными, артериальной гипертензии нет, а объем внеклеточной жидкости снижен, что проявляет себя ортостатической артериальной гипотензией и другими признаками гиповолемии. Кроме того, у больных с этим синдромом выявляют рост экскреции натрия с мочой, метаболический алкалоз и гипокалиемию. У больных с метаболическим алкалозом первого вида его устраняет восстановление объема внеклеточной жидкости внутривенным вливанием изоосмоляльных растворов. Во-первых, такая инфузия восстанавливает массу внеклеточной жидкости, дефицит которой служит стимулом для активации ренин-ангиотензин-альдостеронового механизма как причины метаболического алкалоза. Устранение дефицита объема внеклеточной жидкости увеличивает скорость клубочковой фильтрации, что усиливает бикарбонатурию, компенсирующую метаболический алкалоз. Коррекция дефицита в организме калия нормализует кислотно-основное состояние только в техредких случаях, когда гипокалиемия представляет собой ведущую причину метаболического алкалоза. При такой гипокалиемии содержание К+ в плазме крови меньше, чем 2,4 ммоль/л. Если причиной гиперальдостеронизма (повышенной секреции альдостерона) и низкой скорости клубочковой фильтрации, вызывающих метаболический алкалоз, является застойная сердечная недостаточность, инфузию изоосмоляльных растворов нельзя признать патогенетически обоснованным способом коррекции метаболического алкалоза. У таких больных, кроме кардиотропной фармакотерапии и других лечебных воздействий, направленных на усиление насосной функции сердца, при условии гипокалиемии метаболический алкалоз может устранить внутривенное вливание растворов калия хлорида. Инфузия растворов хлорида калия снимает гипокалиемию, которая снижает скорость клубочковой фильтрации и увеличивает образование почками бикарбонатных анионов. Если метаболический алкалоз второго вида развивается у больных с почечной недостаточностью, как это бывает при реноваскулярной артериальной гипертензии, то концентрация протонов во внеклеточной жидкости и жидкой части плазмы крови падает до уровня более низкого, чем 20 нмоль/л, при рН больше 7,7. Такое значительное возрастание рН связано с неэффективностью при низкой скорости клубочковой фильтрации бикарбонатурии как компенсаторной реакции в ответ на метаболический алкалоз. Метаболический алкалоз такой степени тяжести служит показанием к внутривенному вливанию растворов соляной кислоты или хлорида аммония. Таким больным может быть показан гемодиализ. При этом для уменьшения содержания НСО3- в растворе для гемодиализа снижают содержание бикарбонатных и ацетатных анионов. Когда причина метаболического алкалоза - повышенная секреция минералокортикоидов, то применение лекарственных средств, блокирующих реабсорбцию натрия в собирательных канальцах (амилорид и др.), илиантагонистов альдостерона служит патогенетическим средством коррекции нарушений кислотно-основного состояния. Следует заметить, что все перечисленные способы коррекции метаболического алкалоза не ориентированы на этиологический фактор болезней и патологических состояний, вызывающих метаболический алкалоз. Например, при внутрибрюшном абсцессе какпричине высокой кишечной непроходимости, частой рвоты и метаболического алкалоза нормального кислотно-основного состояния не восстановить без лапаротомии с целью радикальной санации очага гнойной инфекции и устранения непроходимости. |