учебник по патфизу. Учебник для слушателей и курсантов военномедицинской академии и военномедицинских институтов под редакцией проф. В. Ю. Шанина
 Скачать 4.96 Mb. Скачать 4.96 Mb.
|

|
Глава 11. ДИСЛИПОПРОТЕИНЕМИИ И АТЕРОСКЛЕРОЗ Патогенез гиперлипопротеинемии первого типа Семейный дефицит липопротеинлипазы наследуется по аутосомно-рецессивому типу. Так как при данном нарушении липидного обмена в крови растет содержание не связанных с патогенезом атеросклероза триглицеридов и хиломикронов, то с высоким риском атеросклероза данная дислипопротеинемия не связана. Алолипопротеин С-II - это одна из составляющих липопротеинов очень низкой плотности и липопротеинов высокой плотности. Представляет собой эндогенный активатор липопротеинлипазы. При наследственном дефиците аполипопротеина С-II возникает состояние, аналогичное дислипопротеинемии первого типа (синдром Брекенриджа), которое, как и семейный дефицит липопротеинлипазы, в частности характеризует панкреатит, связанный с высокими концентрациями в крови триглицеридов и хиломикронов. Панкреатит данной этиологии может привести ко вторичному сахарному диабету. Патогенез семейной гиперхолестеринемии (дислипопротеинемии второго типа) Семейная гиперхолестерицемия второго типа наследуется по аутосомно-доминантному типу. Она представляет собой результат закрепления в ряду поколений мутаций аллелей Rbo, Rb-b и Rtio. В результате данных мутаций экспрессия рецепторов к липопротеинам низкой площостд на поверхности клеток, образующих холестерин, недостаточна. Низкая экспрессия рецепторов снижает пиноцитоз атерогенных липопротеинов низкой плотности. В результате в данных клетках дадает содержание свободного холестерина, что растормаживает ключевой фермент синтеза холестерина, гидроксиметилглютарил-коэнзим А-редуктазу. Синтез холестерина растет, и нарастает гилерхолестеринемия. Гиперхолестеринемия повышает образование атерогенных липопротеинов низкой плотности гепатоцитами. Кроме того, их содержание в крови растет из-за недостаточного пиноцотоза данных атерогенных липо- протеинов гепатоцитами. Высокая концентрация атерогенных липопротеинов низкой плотности во внеклеточной жидкости и плазме крови обусловливает высокий риск атеросклероза и приводит к инфаркту миокарда в период онтогенеза от 20 до 50 лет. Атерогенность липопротеинов низкой плотности Липопротеины низкой плотности представляют собой субстраты для свободнорадикального окисления с образованием окисленных форм липопротеинов низкой плотности. Окисленные формы липопротеинов низкой плотности - это эндогенные лигандык “рецёпторам-мусорщикам” наружной поверхности эндотелиальных клеток. Их взаимодействие с “рецепторами-мусорщиками” активирует эндотелиальше клетки, которые винактивиройанном состоянии высвобождают факторы клеточного pостa, цитокины-флогогены и свободные кислородные радикалы. Таким образом, индуцируется хроническое воспаление сосудистой стенки с патогенным пролиферативным компонентом, составляющее морфопатогенез возникновения и деструкции атеросклеротйческой бляшки. Вероятность деструкции атеросклеротической бляшки определяет характер воспаления сосудистой стенки. Главным эффектором воспаления являются мононуклеарные фагоциты. Позитивный эффект таких антибактериальных препаратов, как рокситромицин и азитромицин, на атерогенез и нестабильность атеросклеротической бляшки послужили основой дляпредположения о том, что Chlamydiapneumoniaeиграет роль в этиологии атеросклероза. Кроме того, данное действие антибиотиков из группы макролидов связывают с их влиянием на калиевый канал наружной мембраны мононуклеарных фагоцитов, в резудатате чего происходит депрессия макрофагов как клеточных эффекторов воспаления. Острые респираторные инфекции - это фактор риска инфаркта миокарда, внезапной сердечной смерти и нестабильной стенокардии. Гиперлипидемии третьего, четвертого и пятого типов В основе гиперлипидемии третьего типа лежит наследуемая по аутосомно-доминантному типу врожденная недостаточность катаболизма атерогенных липопротеинов промежуточной плотности. Гиперлипидемию третьего типа характеризуют ускоренное развитие атеросклероза и высокий риск тромбоза венечных артерий и острого инфаркта миокарда, сахарный диабет, ожирение, гипотиреоз и особо выраженный ксантоматоз. У больных выявляют poст в плазме крови концентраций триглицеридов и холестерина, которые липопротеины промежуточной плотности переносят от печени на периферию. О сахарном диабете у таких больных свидетельствует сниженная толерантность по отношений к глюкозе. Гиперлипидемия четвертою типа - это наследуемое по аутосомно-доминантному типу нарушение липидного обмена, которое характеризует гипертриглицеридемия, то есть аномально высокое содерние триглицеридов в плазме крови. Это наиболее частое из нарушений: липидного обмена у больных, при котором атеросклероз редко поражает периферические и венечные артерии. При гиперлипидемии четвертого типа в плазме крови повышены концентрации тиглицеридов и липопротеинов очень низкой плотности. Отдельно выделяют приобретенную гиперлипидемию четвертого типа, котоую вызывают сахарный диабет, уремия, препараты из группы глюкокортикоидов, а также бета-адреномиметики. Гиперлипидемия гиперлипопротпеинемия пятого типа - это полиэтиологичное нарушение липидного обмена, вследствие которого у части больных возникает ксантоматоз и панкреатит как следствия патогенно высоких концентраций в плазме крови липопротеинов очень низкой плотности и хиломикронов. Ведущие звенья патогенеза атеросклероза Атеросклеротическое перерождение сосудов во многом определяет пролиферация миоцитов сосудистой стенки, индуцируемая факторами клеточного роста тромбоцитов, активированных мононуклеаров, а также самих гладкомышечных клеток. Термин “атеросклероза” описывает изменения сосудистой стенки при этом системном типовом патологическом процессе. Слово “атере” (греч.) означает “кашица” иуказывает на образование в стенках сосудов кашицеобразных липидных отложений с их дальнейшим склерозированием и уплотнением. По определению Всемирной организации здравоохранения: “Атеросклероз - зто вариабельная комбинация изменений внутренней оболочки (интимы) артерий, включающая накопление липидов, сложных углеводов, фиброзной ткани, компонентов крови, кальцификацию и сопутствующие изменения средней оболочки (медии)”. При гистологическом исследовании пораженных атеросклерозом участков сосудистой стенки выявляют характерное патологическое образование, атеросклеротическую бляшку. Ее формируют липиды, лейкоциты, гладкомышечные клетки и межклеточное вещество интимы артерий. Образованию бляшки предшествует адгезия (прилипание) циркулирующих моноцитов и лимфоцитов к артериальному эндотелию с последующим локальным накоплением пенистых клеток. Необходимым условием адгезии является экспрессия на поверхности эндотелиальных клеток адгезивных молекул (активация эндотелиальных клеток). Пенистые клетки представляют собой активированные мононуклеары, видоизмененные в результате эндоцитоза лицидов. Некоторые липопротеины можно считать атерогенными, неатерогенными, а третьи антиатерогенными. Под атерогенными следует понимать липопротеины, высокая коцентрация которых в плазме крови достоверно связана с развитием атеросклероза. Поверхность макрофагов содержит рецепторы к липопротеинам очень низкой плотности и непостоянные рецепторы к измененным в результате окисления липопротеинам низкой плотности (“рецепторы-мусорщики”). Предположительно активация мононуклеаров черезсвязывание зтих рецепторовс окисленнымиатерогенными липопротеинами ведет к активации субинтимальных мононуклеарныхфагоцитов, в результате которой они приобретают способность к эндоцитозу липопротеинов. В ранней стадии атеросклеротического поражения сосудистой стенки в ней выявляют липидные прожилки и пенистые клетки. Этот начальный этап формирования атеросклеротической бляшки вызывает возбуждение непостоянно присутствующих на клеточной поверхности макрофагов - рецепторов к окисленным липопротеинам низкой плотности (ацетил-рецепторы ЛПНП, “рецепгорн-мусорщики”). Эти рецепторы обладают высоким сродством к окисленным липопротеинам низкой плотности (“мусору”). Изменения липопротеинов низкой плотности в результате окисления, которое обусловливает их высокое сродство к “рецепторам-мусорщикам”, включают:
Как результат патогенных межклеточных взаимодействий атеросклероз представляет собой патологические изменения стенок артерий большого и среднего диаметров, которые составляют и вызывают локальная аккумуляция в интиме липидов и мононуклеаров, миграция и пролиферация гладкомышечных клеток, а также отложения вещества внеклеточного матрикса. Патогенные межклеточные взаимодействия, приводящие к атеросклерозу, реализуются не только через действие факторов роста, но и в результате эффектов цитокинов-флогогенов, медиаторов воспаления и экспрессии адгезивных молекул. Начальным этапом патогенеза атеросклероза как последовательности межклеточных взаимодействий является адгезия моноцита циркулирующей крови к эндотелиальным клеткам с их последующей миграцией винтиму. При этом индукторы атеросклероза, окисленные (модифицированные) липопротеины низкой плотности, воздействуя на лейкоциты циркулирующей крови и эндотелиальные клетки, вызывают экспрессию на их поверхности адгезивных молекул. Так, лизофосфатидилхолин - элемент молекулы окисленных атерогенных липопротеинов - индуцирует экспрессию межклеточной адгезивной молекулы-1 на поверхности эндотелиальных клеток. Известно, что атерогенные липопротеины оказывают на эндотелий прямые и непрямые влияния, повышающие экспрессию на их поверхности эндотелиально-лейкоцитарных адгезивных молекул (ЭЛАМ). Непрямой эффект атерогенных липопротеинов на рост экспрессии таких адгезивных молекул состоит в стимуляций паракринной секреции гладкомышечными клетками и мо-нонуклеарными макрофагами сосудистой стенки. Адгезия лейкоцитов к сосудистой стенке служит первым этапом их проникновения в интиму. Там активированные моноциты секретируют ряд активаторов эндотелия и хемоаттрактантов, стимулирующих дальнейшую инфильтрацию очага атеросклеротического повреждения моноцитами и лимфоцитами из циркулирующей крови, Патогенное функционирование иммунокомпетентных клеток в качестве эффекторов атеросклероза указывает на участие вразвитии атеросклероза иммнопатологической реакции. Атеросклероз - это хроническое воспаление сосудистой стенки, протекающающее с преобладанием пролиферативнго компонента. Его основными клеточными эффекторами являются эндотелиоциты, моноциты циркулирующей крови, мойонуклеарные фагоциты субинтимального слоя, гладагомышечные сосудистые клетки, актвированные окисленными атерогенными липопротеинами или цитокинами-флогогенами. Воспаление в очаге атеросклеротического поражения усиливает взаимосвязанное с ним локальное свертывание крови, которое могут вызвать сами атерогенные липорротеины. Так, атерогенный липопротеин содержит гдикопротеин, связанный р аполипротеином В. Идентичность структуры липопротеина строению плазминогена может обусловливать внутрисосудистый тромбогенез через конкурентное связывание рецепторов к плазминогену на поверхности эндотелиоцитов. Атеросклеротическое перерождение сосудистой стенки начинается с образования прожилок атерогенных липопротеинов и пенистых макрофагов в сосудистой стенке. В данном начальном этапе формирования атеросклеротической бляшки особую роль играют мононуклеарные фагоциты сосудистой стенки, которые сами могут индуцировать окисление липопротеинов низкой плотности через высвобождение свободных кислородных радикалов и (или) активацию липооксигеназы. Так как мононуклеары представляют собой эффектор единой системы иммунитета организма, то мы вправе предположить роль в развитии атеросклероза нервного эндогенного этиологического фактора, столь сильно меняющего состояние системы иммунитета, и ее макрофагального звена в частности. Кроме того, патогенный стресс как состояние, которое характеризует системная интенсификация свободнорадикального окисления липидов, может повышать уровень окисления липопротеинов низкой плотности и тем самым предрасполагать к атеросклерозу. Пенистые клетки высвобождают ряд цитокинов, чье действие вызывает пролиферацию клеточных элементов, вособенности миоцитов гладкомышечных элементов сосудистой стенки. Кроме того, цитокины активированных макрофагов активируют эндотелиоциты, что ведет к росту экспрессии их тромбогенного потенциала. Цитокины пенистых клеток активируют и нейтрофилы циркулирующей крови, что вызывает воспаление с полиморфонуклеарами в качестве его клеточных эффекторов. В результате в составе атеросклеротичсеской бляшки находятся пролиферирующие миоциты сосудистой стенки, агрегаты активированных тромбоцитов и других форменных элементов крови, активированные нейтрофилы и нити фибрина. Все это характеризует атеросклеротическую бляшку как очаг воспаления и локус тромбоза. Макрофаги, окисленные липопротеины, ацетил-ЛПНП-рецепторы играют определяющую роль в индукции атеросклероза. В дальнейшем процесс образования атеросклеротической бляшки теряет связь с этими этиологическими факторами, то есть через патогенные межклеточные взаимодействия происходит его эндогенизация. В 70-е годы были сформулированы основные положения теории атеросклероза, которую назвали теорией “реакция на травму”, В соответствии с данной теорией, механическое и токсическое повреждение эндотелия обнажает субэндотелиальные структуры. В результате обнажения субэндотелиальных структур происходит адгезия тромбоцитов к макромолекулам субэндотелиального слоя. Адгезия активирует тромбоциты, которые вактивированном состоянии высвобождают вещества, стимулирующие тромбообразование: аденозиндифосфат, тромбин, фактор тромбоцитов III. Кроме того, активированные тромбоциты высвобождают фактор клеточного роста тромбоцитов. Высвобождение факторов клеточного роста и тромбогенеза стимулирует пролиферацию гладкомышечных клеток, повышает массу межклеточного матрикса, а также вызывает отложение фибрина и образование тромба в месте повреждения эндотелия, что и формирует атеросклеротйческую бляшку. Современные представления о патогенезе атеросклероза во многом повторяют теории “реакции на травму”. При этомпод травмой следует понимать не морфологический дефект эндотелия, а патогенное изменение функционального состояния эндотелиоцитов. Функциональноесостояние эндотелиальной клетки - это детерминанта образования вазоактивных веществ, строения межклеточного матрикса и секреции факторов клеточного роста на уровне стенки сосудов. Патогенное состояние эндотелиальнойклетки как инициирующий момент патогенеза атеросклероза в основном обусловлено ее реакцией на взаимодействие с окисленными атерогенными липопротеинами низкой плотности. Образование окисленных атерогенных липопротеинов связано с образованием и высвобождением свободных кислородных радикалов. Воздействие свободных кислородных радикалов на атерогенные липопротеины приводит к появлению в их составе окисленных жирных кислот и фрагментации аполипопротеина В-100. В результате таких изменений структуры липопротеина он перестает быть лигандом к рецепopaм липопротеинов низкой плотности и связывается с “рецепторами-мусорщиками” на наружной клеточной поверхности эндотелиоццитов. Это активирует эндотелиальную клетку, которая в активированном состоянии начинает активно высвобождать свободные кислородные радикалы. Кроме того,в активированном состоянии эндотелиальная клетка высвобождает ряд биоактивных веществ, цитокинов, прокоагулянтови факторов клеточного роста. Гиперактивация эндотелиальной клетки через секрецию флогогенов вовлекает в воспаление мононуклеары сосудистой стенки, которые в активированном состоянии становятся клеточными эффекторами воспаления, которое и составляет в основном патогенез атеросклероза (рис.11.1), обусловливая альтерцию сосудистой стенки и образование атероскле-ротической бляшки. В активированном состоянии эндотелиальная клетка репрессирует на своейповерхности адгезивные молекулы. Это стимулирует адгезию мононуклеарных и полиморфонуклеарных фагоцитов к эндотелиоцитам как Начальный этап преимущественно мононуклеарного воспаления, составляющего патогенез атеросклероза. 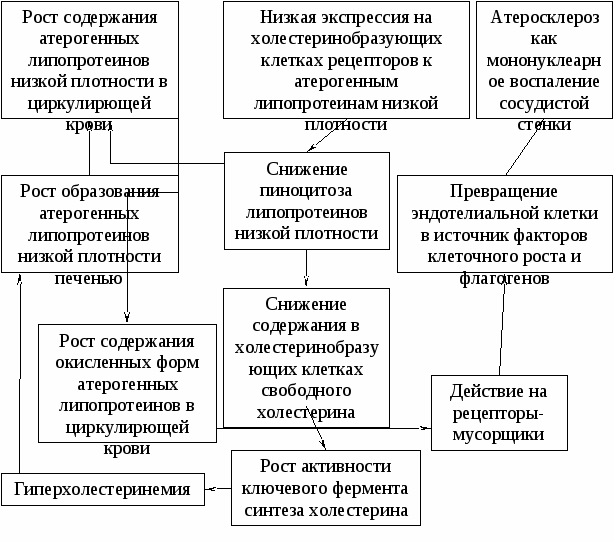 Рис. 11.1. Схема ведущих звеньев патогенеза атеросклероза при пшерли-попротеинемии второго типа Глава 12. РЕАКЦИИ ПОВЫШЕННОЙ ЧУВСТВИТЕЛЬНОСТИ Реакция системы иммунитета в ответ на антигенную стимуляцию может обусловить патологические изменения органов и тканей своего организма. Реакции повышенной чувствительности первого типа Реакция повышенной чувствительности I типа (анафилаксия, от греч. Ana – вновь + phylaxis - беззащитность) возникают при образовании гомоцитотропных антител с высоким сродством (аффинностью) к тучным клеткам и базофилам в ответ на стимуляцию системы иммунитета антигенами, выступающими при генерации способности к анафилаксии в качестве аллергенов, то есть антигенов, взаимодействии с которыми системы иммунитета обуславливает реакцию гиперчувствительности. Когда в начале девятнадцатого века была впервые описана сенная лихорадка, данная патология встречалась весьма редко. В настоящее время один из двух жителей развитых стран страдает от аллергии. Главное в снижении летальности, связанной с критическими состояниями вследствие аллергии, - это не разработка новых способов неотложной терапии. Они известны и в ближайшем будущем изменений не претерпят. Снизить частоту критических состояний, обусловленных реакциями повышенной чувствительности, может лишь активная и патогенетически обоснованная профилактика. В этой связи данная глава в основном будет посвящена современным способам предупреждения реакций повышенной чувствительности как причин критических состояний. В основе большинства из аллергических болезней лежит атопия. В контексте современной иммунопатологии под атопией следует понимать наследственную предрасположенность к образованию и высвобождению иммуноглобулинов E в ответ на действие факторов внешней среды, которые при отсутствии атопии не повышают продукции и содержания в циркулирующей крови данных иммуноглобулинов (гомоцитотропных антител). Иммуноглобулины E как гомоцитотропные антитела обладают способностью сенситизировать тучные клетки во всех органах и тканях. Поэтому спектр аллергических болезней, их проявлений, а также особенностей патогенеза соответствующих критических состояний достаточно широк. Следует заметить, что, несмотря на снижение тяжести аллергических болезней под влиянием современной терапии, риск критических состояний аллергического генеза остается высоким. Атопия наследуется на полигенной основе. Как фенотипические признаки аллергия и атопия могут появляться по ходу онтогенеза по мере накопления в организме последствий влияний на него определенных эндо- экзогенных факторов, то есть условий возникновения аллергических заболеваний. К таковым относят: а) поступление аллергенов с пищей при вскармливании младенцев и питании детей; б) побочное действие антибиотиков, которые особенно часто выступают в качестве аллергенов в детском возрасте; в) эффект вредных примесей к вдыхаемому воздуху (компоненты дыма сигарет и др.); г) действие аллергенов, которые содержит вдыхаемый воздух (антиген березовой пыльцы и др.). |