учебник по патфизу. Учебник для слушателей и курсантов военномедицинской академии и военномедицинских институтов под редакцией проф. В. Ю. Шанина
 Скачать 4.96 Mb. Скачать 4.96 Mb.
|

|
Глава 4. ДИССЕМИНИРОВАННОЕ ВНУТРИСОСУДИСТОЕ СВЕРТЫВАНИЕ Этиология и патогенез Синдром диссеминированного внутрисосудистого свертывания (ДВС) характеризуется системной активацией механизмов гемокоагуляции. Активация механизмов свертывания вызывает образование фибрина в просвете сосудов. Следствием образования фибрина при ДВС является распространенная тромбоэмболия сосудов небольшого и среднего диаметров. Посредством тромбоэмболии ДВС расстраивает микроциркуляцию и лишает органы нормальных структурно-функциональных элементов. Обтурация сосудов вследствие ДВС служит одним из звеньев патогенеза множественной системной недостаточности. Потребление тромбоцитов при действии внешних механизмов свертывания крови, а также потребление при ДВС факторов свертывания вызывают тромбоцитопению и нехватку факторов свертывания (коагулопатию потребления). Коагулопатия потребления обуславливает повышенную кровоточивость, кровоизлияния и массивную кровопотерю. Кровоточивость, кровоизлияния и кровопотеря – это те осложнения ДВС, которые затрудняют лечение, направленное на его устранение (рис. 4.1). 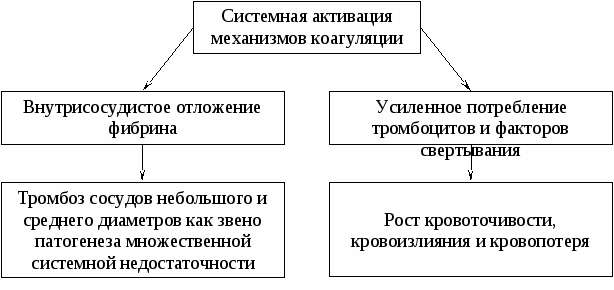 Рис. 4.1. Ведущие звенья патогенеза синдрома ДВС (системная активация коагуляции ведет к распространенному внутрисосудистому отложению фибрина, а также к истощению резервов тромбоцитов и факторов свертывания; в результате возникает тромбоз сосудов небольшого и среднего диаметров, а коагулопатия потребления служит причиной роста кровоточивости, кровоизлияний и кровопотери) Таблица 4.1. Этиология диссеминированного внутрисосудистого свертывания
ДВС – это приобретенное расстройство гемостаза, которое является следствием ряда разных по происхождению болезней и патологических состояний (табл. 4.1). Обычно ДВС обусловлен септицемией. Хотя вызывать ДВС способны практически все патогенные и условно патогенные микроорганизмы, бактериальные инфекции обуславливают ДВС, чаще других. Отчетливые признаки ДВС выявляют у 30-50% больных в состоянии сепсиса, обусловленного инвазией во внутреннюю среду грамотрицательных бактерий. Частота ДВС при грамположительной септицемии находится на том же уровне. Индукторами ДВС при септицемиях являются специфичные компоненты наружных клеточных мембран бактериальных патогенов. К ним относятся липополисахарид или эндотоксин, а также бактериальные экзотоксины (стафилококковый, гемолизин и др.). Компоненты мембран бактерий, эндотоксин, экзотоксины могут индуцировать системную воспалительную реакцию, которую характеризует активация цитокиновой сети. Цитокиновая сеть – это совокупность путей передачи информации между клетками систем врожденного иммунитета, а также иммунокомпетентными клетками посредством высвобождения и действия агентов ауто - паракринной регуляции (цитокинов, то есть движителей клеток). При активации цитокиновой сети эффекты цитокинов трансформируют клетки систем врожденного и приобретенного иммунитета в действующие эффекторы защитных реакций. В данном контексте под клеточными эффекторами следует понимать те клетки, действия которых в определенном функциональном состоянии осуществляют какую-либо физиологическую или защитную реакцию. ДВС – это звено патогенеза необходимо присущее тяжелой раневой болезни и тяжелой травматической болезни мирного времени. Подчеркнем, что чаще всего ДВС возникает при тяжелой черепно-мозговой травме. В остром периоде тяжелой раневой болезни звеньями ДВС являются: 1) высвобождение свободных жирных кислот и фосфолипидов в циркулирующую кровь поврежденными и гипоксичными тканями; 2) гемолиз и повреждения эндотелия в тех же тканях; 3) системное превращение эндотелиоцитов в эффекторы воспаления и коагуляции под действием гиперцитокинемии (следствие активации цитокиновой сети). Активация цитокиновой сети при сепсисе и ее активация при тяжелой сочетанной травме являются идентичными. Частота ДВС у больных с тяжелой сочетанной травмой и системной воспалительной реакцией варьирует от 50 до 70%. Как твердые опухоли, так и злокачественные заболевания крови могут вызывать диссеминированное свертывание. При метастазировании злокачественных опухолей ДВС выявляют у 10-15% больных. При острых лейкозах ДВС идентифицируют у 15% пациентов. В индукции ДВС участвует тканевой фактор свертывания, который содержится на поверхности злокачественных клеток. При ДВС вследствие острой промиелоцитарной лейкемии преимущественно действует такое звено патогенеза ДВС как усиление фибринолиза. У таких больных ДВС в основном проявляется повышенной кровоточивостью. При этом аутопсийные исследования выявляют системный микротромбоз. ДВС-подобное патологическое состояние нередко осложняет микроангиопатические гемолитические анемии. К ним относятся:
Распространенный тромбоз микрососудов, а также сосудов небольшого и среднего диаметров при микроангиопатических гемолитических анемиях в первую очередь является следствием системного повреждения эндотелия. Системное повреждение эндотелия обуславливает адгезию и агрегацию тромбоцитов, образование тромбина и недостаточность фибринолиза. У больных с гемолитическим-уремическим, а также с тромбоцитопеническим синдромом развивается приобретенный дефицит протеазы а. Протеаза а расщепляет сложную молекулу фактора фон Виллебранда на две или более простые. В результате низкой активности протеазы в сосудах накапливаются значительные конгломераты фактора, вызывающие распространенный тромбоз. Патогномоничным признаком микроангиопатических гемолитических анемий служит появление в циркулирующей крови шизоцитов3. Шизоцитоз (аномальный рост содержания шизоцитов в крови), может быть признаком ДВС, обусловленного усилением образования тромбина и действием внешних механизмов свертывания крови. Ведущими звеньями патогенеза ДВС являются:
Сепсис, системная воспалительная реакция и ДВС во многом являются следствиями гиперцитокинемии, то есть патогенного роста содержания в крови провоспалительных цитокинов (фактора некроза опухолей-альфа, интерлейкина-1 и др.). Главным активатором механизмов свертывания крови при ДВС является интерлейкин-6. Фактор некроза опухолей-альфа (ФНО) не обладает прямым действием на свертывание крови. Как фактор коагуляции ФНО влияет на систему интерлейкина-6, являясь ведущим медиатором нарушений регуляции физиологических механизмов, действие которых противостоит свертыванию крови. Одновременно (ФНО) угнетает физиологическую систему фибринолиза. Системный рост образования тромбина как ведущее звено патогенеза ДВС происходит исключительно посредством активации внешних механизмов гемостаза, которые действуют с участием тканевого фактора свертывания крови и активированного седьмого фактора (VIIa) (рис. 20. 2). Угнетение активности тканевого фактора или VIIa полностью устраняет образование тромбина и ДВС, вызванные действием эндотоксина. Таким эффектом не обладает блокада действия внутренних (присущих самой крови и ее плазме) механизмов гемокоагуляции. При сепсисе и системной воспалительной реакции мононуклеарные фагоциты экспрессируют тканевой фактор свертывания в ответ на действие провоспалительных цитокинов. Не исключено, что также ведут себя эндотелиальные клетки. Одним из ведущих звеньев патогенеза ДВС является недостаточность действия ингибиторов свертывания крови. При ДВС нарушены действия механизмов всех главных физиологических противосвертывающих систем: 1) антитромбина III; 2) белка C; 3)ингибитора тканевого фактора свертывания крови. У больных с диссеминированным свертыванием в плазме крови выявляют аномально низкую концентрацию антитромбина III, главного антагониста тканевого фактора на путях каскадов реакций свертывания. Причинами патологически низкого содержания антитромбина III являются: 1) его утилизация при ДВС; 2) разрушение антикоагулянта эластазой, высвобождаемой нейтрофилами, активированными для воспаления; 3) снижение синтеза антитромбина III. Угнетение системы протеина C - это следствие низкого образования данного белка, снижения активности эндотелиального тромбомодулина под действием провоспалительных цитокинов, а также прогрессирующего снижения содержания в плазме крови белка S (кофактора протеина C при их совместном противосвертывающем действии). Действие тканевого фактора – это ключевой момент индукции ДВС. Действиям тканевого фактора на путях каскадов реакций свертывания крови противостоят эффекты ингибитора тканевого фактора свертывания крови (ИТФ). Несмотря на то, что при ДВС не выявлены врожденные и приобретенные расстройства системы ИТФ, отсутствие нормальных влияний ИТФ на свертывание крови можно считать звеном патогенеза диссеминированного свертывания. Угнетение фибринолитической активности – одно из звеньев патогенеза ДВС. При ДВС в крови фиксируют повышенную активность ингибитора активатора плазминогена первого типа (ИАП I). Напомним, что ингибитор является главным медиатором торможения фибринолитической системы. 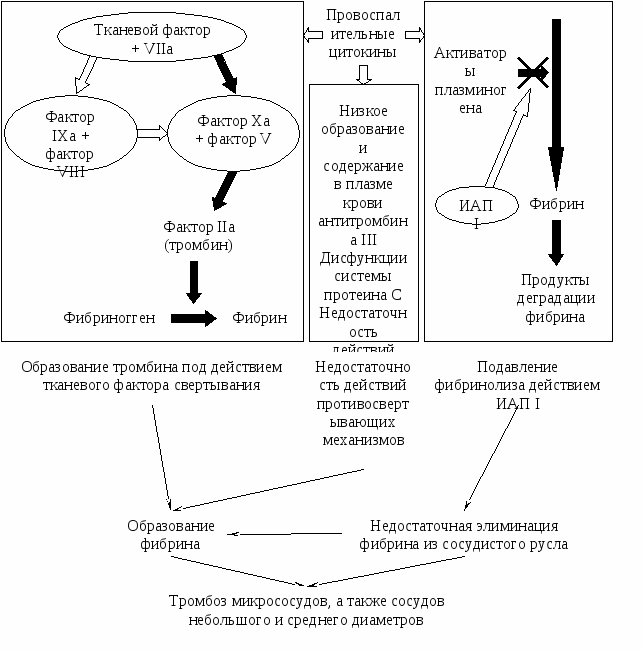 Рис. 4.2. Ведущие звенья патогенеза ДВС (при ДВС фибрин образуется под действием тканевого фактора свертывания крови; тканевой фактор (ТФ) под действием провоспалительных цитокинов появляется на поверхности мононуклеарных фагоцитов и эндотелиальных клеток, ТФ связывает и активирует фактор VII; комплекс ТФ и активированного фактора VII (VIIa) активирует фактор Х прямо (черная стрелка) и опосредованно («прозрачные» стрелки), то есть через активацию фактора IХ и фактор VIII. Активированный фактор Х вместе с фактором V действуют на протромбин. В результате образуется тромбин. Одновременно происходят расстройства трех главных противосвертывающих систем: антитромбина III, белка C, а также ИТФ. Это усиливает образование фибрина при его недостаточной элиминации из сосудистого русла фибринолизом. Фибринолиз подавляется усиленным действием ИАП I, которое, угнетая активность активаторов плазминогена и снижая образование плазмина, угнетает деградацию фибрина. Под действием звеньев патогенеза ДВС образование фибрина (Ф) преобладает над его элиминацией из сосудистого русла. Избыточное образование Ф служит причиной распространенного микротромбоза). Патогенетические принципы лечения ДВС Интенсивная терапия ДВС неэффективна, если обратному развитию под влиянием системы лечения не подвергается патологическое состояние или болезнь, которые являются первичными по отношению к ДВС. На содержание терапии ДВС влияет превалирование того или иного звена его патогенеза. Терапия при преобладании коагулопатии потребления как причины патологической кровоточивости отличается от лечения при системном тромбозе, обусловленном ДВС . Действие гепарина может ослабить свертывание крови, патологически усиленное сепсисом и системной воспалительной реакцией. Кроме того, действие гепарина при ДВС используется с целью предупреждения тромбоза глубоких вен нижних конечностей и таза. Следует заметить, что действие гепарина как антикоагулянта может быть опасным при коагулопатии потребления, обусловленной ДВС. Использование гепарина можно считать оправданным, когда есть признаки преобладания тромбоза сосудов небольшого и среднего диаметров над коагулопатией потребления (молниеносная пурпура и др.). В таких случаях начинают непрерывную внутривенную инфузию раствора гепарина со скоростью 300 – 500 единиц в час. Рациональной альтернативой является введение фракционных доз низкомолекулярных гепаринов. В настоящее время все шире используют ингибиторы тромбина, не действующие на антитромбин-III, то есть дисрудин и другие лекарственные средства. Эти препараты пока не могут заменить гепарин. Дело в том, что их действие связано с более высоким риском кровотечений. При коагулопатии потребления дефицит тромбоцитов и коагулянтов в крови больного устраняют посредством трансфузий тромбоцитарной массы и плазмы. Для обратного развития коагулопатии потребления, которую обуславливает ДВС, необходима инфузия не менее шести международных единиц плазмы крови за 24 ч интенсивной терапии. Опасность трансфузий тромбоцитарной массы состоит в том, что данный препарат крови содержит следы активированных факторов свертывания крови, эффект которых может обострить коагулопатию потребления. Тромбоцитарная масса содержит только определенные коагулянты, тогда когда у больных с ДВС с кровью в недостаточном количестве циркулируют все факторы свертывания крови. Одной из целей интенсивной терапии, направленной на устранение ДВС, является восстановление физиологических отношений в системе свертывания крови. Напомним, что у пациентов с ДВС всегда в патогенезе действует недостаток в плазме антитромбина III. В настоящее время в ходе клинико-экспериментальных исследований изучают эффективность препаратов антитромбина III в интенсивной терапии ДВС. До сих пор нет свидетельств того, что массивная трансфузия донорской плазмы снижает риск коагулопатии потребления при начавшемся ДВС. Трансфузия препаратов концентратов факторов свертывания противопоказана при ДВС. Дело в том, что такие препараты могут содержать активированные факторы свертывания, действие которых может усилить ДВС и вторичную коагулопатию потребления. Кроме того, следует учитывать, что в настоящее время доступны концентраты какого-либо одного фактора, а ДВС обуславливает дефицит всех факторов свертывания крови. Одно из звеньев патогенеза ДВС – это приобретенный дефицит антитромбина III. Не исключено, что эффекты препаратов на основе концентратов антитромбина III могут снизить тяжесть ДВС и улучшить исходы при патологических состояниях, его обуславливающих (сепсис, тяжелая раневая болезнь), если их применять в стадии гиперкоагулемии при диссеминированном свертывании. Действие антифибринолитических средств может усилить синдром множественной системной недостаточности, связанный с ДВС, звеном патогенеза которого является отложение фибрина на сосудистой стенке микрососудов, а также их распространенный микротромбоз. Не исключено, что в будущем средствами предупреждения ДВС будут являться (Levi M., ten Gate H., 1999):
Глава 5. ГИПОКСИЯ На Земле жизнь возникла в результате прогрессивного развития углеродных соединений, органических веществ, формировавшихся из надмолекулярных, а в дальнейшем более сложных систем. Эволюция живого тесно связана с появлением и наполнением в атмосфере земли кислорода. До возникновения фотосинтеза парциальное давление кислорода в атмосфере составляло только 0,15 мм рт. ст. Кислород - скорее следствие, чем условие возникновения жизни на Земле. Он стал совершенно необходим для большинства видов живых организмов. Сейчас аэробиоз уже не может полностью замениться анаэробным типом обмена («... ан и аэробиоз не являются путями выбора для получения энергии. Это совершенно различные способы жизни», - Сент-Дьёрди 1964). Проблема гипоксии рассматривается в науке уже более 300 лет. Кислород научился получать ещё изобретатель подводной лодки Корнелий Дреббель (1574-1634), великий голландец, проработавший большую часть жизни придворным механиком в Англии. Однако, его работы, были засекречены (как и многие последующие, относящиеся к рассматриваемой проблеме),и принципиальное открытие кислорода произошло во второй половине 18 века шведом К.В. Шесле и англичанином Джозефом Пристли. Вернее сказать, они «описали, но не догадались, что описывают», - как позднее писали классики. Более 200 лет назад А. Лавузье ввел термин «кислород» для обозначения «дефлогистированного воздуха» Джозефа Пристли. Определённую роль кислорода прояснили работы М.В. Ломоносова. Понятие «гипоксия» (происходит от греч. hypо - мало, oxia - кислород) - типический патологический процесс, возникающий в результате недостаточного снабжения тканей кислородом или нарушения использования его тканями. С развитием представлений о гипоксии, естественно, изменилась и терминология. Вначале существовал термин «аноксия» (замененный по предложению Виггерса термином «гипоксия» в 1940 году), который означал все те состояния организма, при которых по тем или иным причинам изменялись параметры доставки кислорода к тканям. Начальник кафедры патофизиологии Медико-хирургической академии В.В. Пашутин внес значительный вклад в изучение механизмов развития гипоксии. В 1880-1890 гг. классифицируя гипоксию, он выделял 2 группы гипоксических расстройств:
К первой группе относились гипоксические состояния, возникающие в результате снижения парциального давления кислорода во вдыхаемом воздухе. Эндогенная гипоксия характеризовалась нарушением доставки кислорода к тканям. Ученик В.В. Пашутина и его преемник П.М. Альбицкий расширил представления о кислородном голодании. В 1905 году он предложил различать виды кислородного голодания в зависимости от причин, которые его вызывают:
В 1922 году М. Питерс и Ван Слайк в понятие гипоксия (в тот период использовался термин «аноксия») как и П.М. Альбицкий включили «гистотоксическую аноксию», то есть состояние, при котором доставка кислорода к тканям не изменена, но нарушена его утилизация. Введение термина «тканевая гипоксия» вызвала значительные дискуссии, которые не прекращаются и до настоящего времени. Существенный вклад в изучение механизмов гипоксических расстройств внесли И.Р. Петров и его сотрудники. Гипоксия (hypoxia; гип- + лат. охуgеniит, кислород) - типовой патологический процесс, который вызывают недостаточное поступление кислорода в ткани и клетки организма или нарушения его использования при биологическом окислении. Гипоксия всегда приводит к недостатку свободной энергии, гипоэргозу. |