Биохимия тканей. Учебное пособие Чита, 2015 ббк 28. 07252. 5 Удк 577. 1 616008 Никитина Л. П., Гомбоева А. Ц., Соловьева Н. В
 Скачать 2.39 Mb. Скачать 2.39 Mb.
|

|
2.4.4. Этапы образования гемоглобина Синтез гемоглобина, как любого другого белка, требует наличия матрицы (иРНК), которая продуцируется в ядре. Эритроцит, как известно, не имеет никаких органоидов; следовательно, формирование гемовых протеинов возможно лишь в клетках-предшественниках (эритробластах, заканчиваясь в ретикулоцитах). Этот процесс у эмбрионов осуществляется в печени, селезёнке, а у взрослых – в костном мозге плоских костей, в которых кроветворные стволовые клетки непрерывно размножаются и генерируют предшественников всех типов клеток крови (эритроцитов, лейкоцитов, тромбоцитов). Формирование первых регулируется эритропоэтином почек. Параллельно с генезом глобина происходит образование гема, облигатным компонентом которого служат катионы железа. 2.4.4.1. Обмен железа Общее содержание железа в организме не превышает 4-5 г, локализуясь в основном в гемопротеидах, в первую очередь в гемоглобине. Поэтому необходимо кратко остановиться на судьбе ионов этого металла в организме (Рис. 2.3). Пищевое железо может иметь различные формы: восстановленную и окисленную. Наиболее хорошо всасывается первая в составе гемина из животных продуктов, которая получается в кишечнике после встречи с витамином С и другими восстановителями. Отсюда диеты, богатые мясом, сводят вероятность экзогенного железодефицита к минимуму. В растительных, особенно зерновых продуктах, до 60% ионов переходного металла находится в трудно усвояемой форме, связанной с фитиновой кислотой. 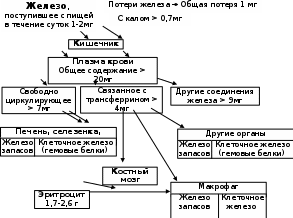 Рис. 2.3. Метаболизм железа в организме Следует заметить, что биодоступность железа невелика: абсорбируется в тонком кишечнике не более 10% содержащегося в пище, в основном животного происхождения. Особую роль при этом играют нормальная секреция соляной кислоты, протеаз, характер питания (способствуют его усвоению сукцинат, лактат, аскорбат), биогенные эффекторы (эритропоэтин из почек стимулирует абсорбцию железа). В энтероцитах ферроионы связываются с апоферритином с образованием ферритина (Fe2+). Попавшие по venaporta в печень ионы металла включаются в гликопротеид трансферрин, с помощью которого транспортируются и путём эндоцитоза проникают в цитозоль клеток-мишеней. В эритроидных клетках железо распределяется между митохондриями, где включается в гем, и белком ферритином, а в миелоидных – его существенная часть попадает в защитный белок – лактоферрин. Если происходит внутрисосудистый гемолиз, то высвободившийся при этом гем, попадая в плазму, связывается с гемопексином (это необходимо вследствие прооксидантных свойств данного порфина) и в таком виде транспортируется в печень, где распадается. После чего ионы железа или вновь используются, или запасаются (Рис. 2.3). Основным депо этого металла служит ферритин, который накапливается в печени, селезёнке, костном мозге, в меньшей степени – в мышечной ткани. Избыток железа может аккумулироваться в выше перечисленных органах также в составе гранул гемосидерина – нерастворимого производного ферритина; железо в нём менее доступно для использования в гемопоэзе. Катаболическая фаза обмена ионов железа заключается в их выведении, в основном в составе желчи через желудочно-кишечный тракт (за сутки в среднем организм теряет до 1,5 мг). Нормой для здорового взрослого человека считается содержание железа в крови 12-30 мкмоль/л. 2.4.4.2. Синтез гема Гем синтезируется практически во всех клетках, кроме эритроцитов. Его образование происходит на ранних стадиях формирования в проэритробластах, эритробластах, ретикулоцитах. Исходными метаболитами процесса служат сукцинил КоА и глицин (Рис.2.4). Сукцинил Ко-А продукт не только ЦТК, но образуется и при распаде треонина, метионина, тимина, ВЖК с нечётным числом атомов углерода, а глицин можно получить из серина с помощью ТГФК. 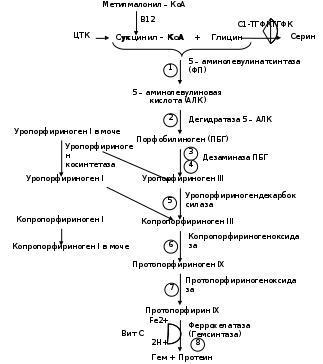 Рис. 2.4. Система ферментов и основные этапы биосинтеза гема Рис. 2.4. Система ферментов и основные этапы биосинтеза гемаВ митохондриях при участии 5-аминолевулинатсинтазы в ходе альдольной конденсации сукцинил-КоА и глицина образуется 5-аминолевулиновая кислота (АЛК). Затем из молекул АЛК, вышедших в цитоплазму, под влиянием специфической дегидратазы (2) синтезируются 4 молекулы порфобилиногена (ПБГ), из которых формируются тетрапиррольные молекулы уропорфириногенов (УП-генов). Данный этап катализируется двумя энзимами: ПБГ-дезаминазой (3) и уропорфириноген-косинтазой (4).В этих условиях обычно возникает изомер III. При отсутствии косинтазы или снижении её активности получается уропорфириноген I, который ограничен в своих дальнейших преобразованиях и, как побочный метаболит, выделяется из организма, а при патологических состояниях и генетических нарушениях может накапливаться (Рис. 2.4). Дальнейшие превращения УП-гена III, молекула которого содержит 8 карбоксильных групп, протекают под контролем УП-гендекарбоксилазы (5), осуществляющей последовательное декарбоксилирование соединения до копропорфириногена III(КП-ген). Затем, после возвращения в митохондрии, это вещество подвергается действию системы энзимов: КП-ген- и протопорфириноген-оксидаз (6,7). Под влиянием первого происходит окислительное декарбоксилирование КП-гена III до протопорфириногена IX, за окисление которого отвечает второй фермент. В клетках костного мозга за сутки синтезируется до 30 мг протопорфирина IX, который комплексируясь с ионами двухвалентного железа (ферроионами), образует гем. Этот этап катализируется феррохелатазой (гемсинтетазой) (8). Гем включается в различные гемопротеины (гемоглобин, миоглобин, каталазу, пероксидазу, цитохромы). 2.4.5.Болезни анаболизма гемоглобина Так как на долю гемоглобина приходится около 30% массы эритроцита, то естественно, что повреждения в его структуре сказываются на продолжительности жизни и функционировании красных кровяных телец. Отсюда выделяют следующие причины дезоксигенации тканей. Нарушения в синтезе Hb, разделённые на: а) сдвиги в метаболизме железа; б) изменения в синтезе гема; в) последствия мутаций в генах, ответственных за образование глобина. Дисгемоглобинемии – диспропорции в балансе различных форм гемопротеидов. Повреждения транспорта плазменного гемоглобина. 2.4.5.1.Анемии как следствие нарушений обмена железа Любая патология в обмене железа сопровождается развитием одного из самых распространённых недугов - анемией, которая обычно характеризуется или эритропенией, или снижением уровня гемоглобина. Общие лабораторные признаки подобных патологических состояний: концентрация Hb менее 100 г/л; число эритроцитов не достигает 4*1012 /л; содержание железа в сыворотке крови ниже 12 мкмоль/л. С практической точки зрения основной и обязательной характеристикой анемии является обеднение гемоглобином, что уменьшает кислородную емкость крови, приводит к гемической гипоксии, с чем и связаны клинические признаки расстройства жизнедеятельности у больных. К симптомам анемии (слабость, вялость, повышенная утомляемость, падение работоспособности, одышка, сердцебиение, тахикардия, раздражительность, ослабление памяти и внимания, головокружение, шум в ушах, бледность кожи и слизистых оболочек, снижение аппетита) присоединяется сидеропенический синдром: изменения кожи, волос, ногтей, кариес зубов; жжение в языке, атрофия его сосочков, гингивит, хейлит, глоссит, снижение вкусовых ощущений, «географический язык», трещины, заеды в углах рта, затруднение при глотании, отрыжка, тошнота, извращение вкуса и обоняния (picachlorotica – употребление несъедобных веществ: мела, угля, песка, глины), дефекты иммунитета. Основным маркером анемии служит концентрация гемоглобина, обеспечивающего доставку О2 тканям. Границей, разделяющей норму и патологию, принято считать количество этого белка <110 г/л. Число эритроцитов – менее информативный критерий и не всегда коррелирует с уровнем снижения величин гемоглобина. Анемии разнообразны по этиологии, патогенезу и клинико–гематологическим особенностям. Основным фактором, провоцирующим развитие заболевания, служат нарушения в обмене железа, причины которых многолики: недостаток железа в питании (у вегетарианцев, при голодании, при использовании разных диет для похудения, при повышенной потребности в нём у беременных, при кормлении грудью, у быстро растущих подростков); при сдвигах в балансе питательных веществ (при недостатке аскорбата, сукцината, избытке фитатов, клетчатки); следствие нарушения всасывания в ЖКТ (при гиперсекреции соляной кислоты, протеаз, после субтотальной гастрэктомии); при повреждениях слизистой кишечника (при язвенной болезни, диафрагмальной грыже, гельминтозах, язвенном колите, после лечения салицилатами, стероидами); из-за обеднения запасов железа (при угнетении активности ферментов синтеза гема); после усиленного выделения ионов железа из организма (при острых и хронических кровопотерях, при геморрое, различных язвах в желудке, кишечнике, повторных эпизодах кровохарканья). В зависимости от степени сохранения количества железа в организме выделяют Fe-дефицитные, Fe-достаточные, Fe-избыточные анемии. Около 90-92% всех случаев подобных заболеваний приходится на первый вариант. В основе других видов анемии лежат нарушения, связанные с неэффективным эритропоэзом (дисгемопоэзом), талассемией, атрансферринемией, гемоглобинозами. В результате не включённые в гем ионы железа начинают откладываться в виде гемосидерина (наследственные и приобретённые гемохроматозы) в органах и тканях (печени, поджелудочной железе, миокарде, суставах, коже) с последующим угнетением их функций. Прослеживаются следующие признаки: цирроз печени, сахарный диабет, бронзовая окраска кожи (бронзовый диабет), инфаркты миокарда и селезёнки. А так как параллельно развиваются симптомы анемии (из-за дефицита гемоглобина в эритроцитах), особенно опасно в этих случаях применение в качестве терапевтических средств препаратов железа. 2.4.5.2. Порфирии Заболевания, в основе которых лежат повреждения в синтезе гема, названы порфириями. Их развитие чаще всего связано с наследственными дефектами в ферментных системах, ответственных за анаболизм порфиринов. Некоторые клинические разновидности подобных патологий считаются приобретёнными, т.к. у больных на первый план в качестве вероятных провоцирующих факторов выступают различные интоксикации. Таблица 2.1 Локализация повреждений ферментов при различных порфириях и их следствие
Примечание:АЛК –аминолевуленовая кислота; П – печёночная; Э – эритропоэтическая; АР – аутосомно- рецессивный, АД – аутосомно-доминантный типы наследования В зависимости от локализации нарушений различают эритропоэтические (аномалии в обмене порфиринов в костном мозге) и печёночные (похожие сдвиги в гепатоцитах) типы (Табл. 2.1), отличающиеся друг от друга дефектами разных генов. Порфирии встречаются во всех странах с различной частотой, достигающей местами очень высокого уровня (острая перемежающаяся порфирия (ОПП) в Лапландии – 3:1000, порфирия Variegata (ВП) в ЮАР – 1:1000). Впервые порфирия (точнее её наследственная эритропоэтическая форма) была описана Шульцем (1874) и Гюнтером (1911). Однако исторические хроники времён средневековья сохранили описания семейств, у членов которых отмечались черты, свойственные тяжелым формам этого страдания, проявляющиеся кожными, неврологическими, психическим и абдоминальными симптомами (клиникой острого живота, тошнотой, рвотой, задержкой стула; эпилептическими припадками, парезами, параличами, полиневритами, зрительными и слуховыми галлюцинациями, слепотой), а также аномально высоким выделением порфинов с мочой или калом. Некоторые признаки болезни – красный оттенок зубов и костей, своеобразный цвет кожи, изменённой волдырями, язвами и рубцами; ночной образ жизни, обусловленный фотодерматитом, спонтанное свечение некоторых тканей и выделений больного, прихоти вкуса, связанные с анемией, – столь ярки и необычны, что вызывают в памяти описание облика и поведения мифических вурдалаков или вампиров. В России чаще выявляются ОПП и ПКП. Для первой характерен комплекс следующих симптомов: тахикардия, повышение АД, коликообразные боли в животе, признаки полиневрита, судороги, изменения поведения, эмоциональная лабильность, депрессия, галлюцинации, парез дыхательной мускулатуры и др. Способы диагностики основаны на выявлении избыточного количества 5-АЛК, порфобилиногена и общих порфиринов (Табл. 2.1). Поздняя кожная порфирия (ПКП) проявляет себя повышенной светочувствительностью, глубокими поражениями кожи, обусловленными фотосенсибилизацией, иногда клиникой «острого живота». При данной форме рост величин общих порфиринов, 5- АЛК, порфобилиногена незначителен (в 1,5- 2 раза), но в крови и моче можно обнаружить порфириногены, абсолютно отсутствующие у здоровых лиц. Иногда острое или хроническое отравление тяжёлыми металлами провоцирует клиническую картину пароксизма порфирий. Ионы этих металлов, взаимодействуя с сульфгидрильными группами аминолевулинатсинтазы или феррохелатазы, подавляют их активность в синтезе гема. В результате в эритроцитах накапливается протопорфирин, плазме крови увеличивается содержание железа, оно откладывается в органах и тканях, провоцируя формирование гемосидероза. В моче повышается экскреция 5-аминолевулината. 2.4.5.3.Гемоглобинопатии Е  стественно, что патология синтеза глобина носит наследственный характер. Выделяют две основные формы нарушений: одиночные аминокислотные повреждения в структуре белка (гемоглобинозы) и подавление выработки в целом какой-либо полипептидной цепи в мицелле (талассемии) (Рис. 2.5). стественно, что патология синтеза глобина носит наследственный характер. Выделяют две основные формы нарушений: одиночные аминокислотные повреждения в структуре белка (гемоглобинозы) и подавление выработки в целом какой-либо полипептидной цепи в мицелле (талассемии) (Рис. 2.5). 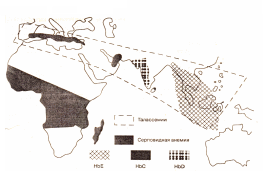 Рис. 2.5. Особенности медицинской географии серповидно- клеточной анемии и других гемоглобинопатий В настоящее время описано более 700 видов патологических гемоглобинов. Первые модифицированные гемопротеиды назывались помощью латинских букв (C, D,E, M, S), но когда число изменённых Hb превысило количество знаков в алфавите, их стали именовать по месту открытия (Kansas, Boston, San Yose, Нirosima, Richmond, Volga и др.). Неоднородный характер генетических дефектов (замена, вставка, сдвиг рамки, удлинение цепи и т.д.) приводит к различным последствиям (изменяется сродство к кислороду, снижается стабильность эритроцитов, что проявляется усиленным гемолизом, цианозом). Это определяется не только выраженностью аномалий в нуклеотидной последовательности, но и природой сменившихся аминокислот. Если же строение мутированного участка меняется резко, то высока вероятность развития серьёзных клинических признаков. Особенно опасны повреждения, локализующиеся в контактных площадках (местах связывания субъединиц в мицеллу) или в карманах, где располагается гем. В этих случаях нарушается формирование гетерогенного тетрамера, что снижает жизнеспособность эмбриона и повышает вероятность выкидыша. Примером может служить HbPhilly, в нём фенилаланин встаёт на место тирозина, который в нормальном белке удерживает отдельные протомеры. После подобной мутации существование генетически изменённого протеина становится невозможным – мицелла разваливается. В гемоглобине Genowaиз-за утери валина происходит смещение фрагмента, остаток глутамата оказывается внутри мицеллы, участок деформируется, что снижает сродство к кислороду, развивается цианоз. Более благоприятные последствия возможны, если мутации сказываются на аминокислотах, создающих поверхность мицеллы. Классическим примером может служить HbS. В нём в β-цепи глутамат замещён валином, т.е. кислое соединение сменяется гидрофобом. Так как повреждение находится снаружи молекулы, то подобное изменение уменьшает заряд и растворимость Hb, а отдельные его мицеллы, сталкиваясь, склеиваются за счёт образования между валинами гидрофобных взаимодействий. Подобная агрегация удлиняет нити гемопротеида, что приводит к их линейной кристаллизации. Это явление сказывается на форме эритроцитов: они принимают форму серпа (Sicklecell – серповидная клетка), теряют эластичность, способность к упругой деформации. Отсюда заболевание носит название серповидной анемии, или гемоглобиноза S. Повреждённая при этом мембрана форменного элемента обладает меньшей устойчивостью, что провоцирует гемолиз, тромбозы. Продолжительность жизни красных кровяных телец сокращается до 15 - 20 суток (норма – 120), что приводит к анемии. Для больных характерны гемолитические кризы с острым болевым синдромом, симптомы поражения селезёнки, печени, ЖКТ, миокарда и др. Частота присутствия подобного гемопротеида в США составляет 8-9% (у афроамериканцев), в некоторых регионах Греции достигает 40%. В течение первого года жизни в Африке 50% детей, больных серповидноклеточной анемией, умирает. Во всём мире более 3 млн. людей страдают талассемиями (thalassa–море) – наследственными заболеваниями, в основе которых лежит репрессия гена, ответственного за синтез какой-либо цепи глобина. При этом формируются мицеллы, часто представляющие собой гомогенные тетрамеры, например, включающие только β- или γ-цепи. HbH, состоящий из 4 β-субъединиц, способен кислород лишь связывать, а отдавать – нет. При угнетении продукции α-цепей (HbBart) гомозиготный зародыш нежизнеспособен, происходит выкидыш на ранних сроках беременности. Встречается в Индокитае. Ребёнок с β-талассемией (болезнью Cooly) (блоком генеза β-цепи) рождается практически здоровым (ведь для роста и развития плода необходим гемоглобин, включающий альфа- и гамма-цепи). Но с каждым месяцем начинают проявляться патологические признаки, определяемые степенью гипоксии, усилением ПОЛ, повреждением мембран эритроцитов [тяжёлая гемолитическая анемия, желтуха, сохранение высокого содержания Hb F (до 20-30%), нарушение физического и психического развития, ребёнок отказывается от груди, лицо приобретает монголоидные черты – скулы выдаются вперёд, основание переносицы вдавливается, нос становится приплюснутым]. Постепенно формируются гепатоспленомегалия, гемохроматоз. Для более старшего возраста характерны гемолитические кризы, лихорадочное состояние, возможно развитие сердечной недостаточности. Лабораторные признаки: гипербилирубинемия (за счёт свободной формы), рост величин плазменного железа. В США введён скрининг на определение патологического гена. Распространение этих заболеваний зависит от региона (Рис. 2.5). Особенно часто они встречаются по берегам морей (Средиземного, Чёрного). В Закавказье регистрируется до 10% носителей таких генов. Ранее талассемии были редки для России, но в связи с меняющейся демографией, миграцией лиц, для которых свойственна данная патология, следует ожидать роста количества детей с этим диагнозом. |