нев. инт 13 2. Выделить основные аспекты диагностики и лечения миастении и миастенических синдромов
 Скачать 97.71 Kb. Скачать 97.71 Kb.
|

ОПРЕДЕЛЕНИЕ НАСЛЕДСТВЕННЫХ МИОТОНИЧЕСКИХ СИНДРОМОВ. КЛАССИФИКАЦИЯ.Наследственные миотонические синдромы(НМС)- группа генетически гетерогенных заболеваний ионных каналов хлора и натрия (каналопатии), характеризующиеся повышенной возбудимостью мембраны мышечных волокон, проявляющиеся миотоническими феноменами с постоянной или транзиторной слабостью скелетной мускулатуры. В современной классификации НМС представлены различными генетическими формами дистрофических (ДМ) и недистрофических миотоний (НДМ) (Mankodi A.2008г) 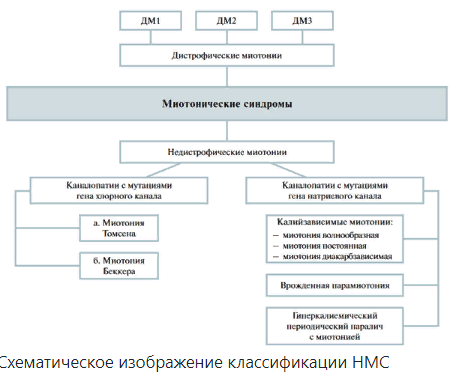 ДИСТРОФИЧЕСКАЯ МИОТОНИЯ. ГЕНЕТИЧЕСКИЕ АСПЕКТЫ, ЭТИОПАТОГЕНЕЗ, КЛАССИФИКАЦИЯДистрофическая миотония (ДМ) (сongenital myotonic dystrophy) является наследственным мультисистемным заболеванием, при котором мутация затрагивает развитие и функционирование различных органов и тканей: гладкой и скелетной мышечной ткани, сердца, органа зрения (глаза), головного мозга. Клиническая картина дистрофической миотонии складывается из 3 синдромов: миотонического, дистрофического и синдрома вегетативно-трофических нарушений. Ключевая особенность заболевания — сочетание миотонии, которая характеризуется отсроченным расслаблением после мышечного сокращения, и прогрессирующей мышечной слабости, дистрофии (атрофии). До 1994 года дистрофическая миотония считалась однородным заболеванием. Однако в последние годы после идентификации различных мутаций при сходной клинической симптоматике, напоминающей дистрофическую миотонию, было показано, что это гетерогенное заболевание, представленное тремя подтипами: дистрофическая миотония 1 типа (мутация 19q13.3), дистрофическая миотония 2 типа (мутация 3q21) и дистрофическая миотония 3 типа (мутация 15q21-q24). Распространенность дистрофической миотонии 1 типа в больших популяциях около 1:8000 , а дистрофической миотонии 2 и 3 типов в настоящее время недостаточно изучена. В отличие от большинства других наследственных нервно-мышечных заболеваний (и от других форм миотоний, в частности) клинические проявления дистрофической миотонии вариабельны и могут различаться от человека к человеку даже в пределах одной семьи. Больные с дистрофической миотонией могут сталкиваться с большим числом разнообразных (необычных для миотонии) проблем, к которым можно отнести низкий уровень жизненной активности, пассивность, депрессию, облысение, нарушения со стороны желудочно-кишечного тракта, сексуальные проблемы, что в ряде случаев приводит к возникновению недопонимания и даже к конфликтным ситуациям между пациентом и лечащим врачом. Следует помнить, что пассивное поведение пациента является по большей мере реальным проявлением данного заболевания, а не желанием человека. КЛАССИФИКАЦИЯ По локализации мутированного гена: 1. дистрофическая миотония 1 типа (мутация 19q13.3) DM1, миотония Штейнерта — Баттена — Куршманна (Steinert — Batten — Curschmann myotonic dystrophy), атрофическая миотония Россолимо; 2. дистрофическая миотония 2 типа (мутация 3q21) DM2; 3. дистрофическая миотония 3 типа (мутация 15q21-q24) DM3; Классификация дистрофической миотонии 1 по возрасту дебюта заболевания: 1. конгенитальная дистрофическая миотония(врожденная, congenital myotonic dystrophy CmyD) — с характерной клинической симптоматикой сразу при рождении ребенка и серьезным прогнозом (не описана при DM2 и DM3); 2. ювенильная дистрофическая миотония(juvenile DM)— возраст дебюта заболевания от 1 года до подросткового возраста; 3. дистрофическая миотония взрослых(adult DM) — с дебютом у индивидуумов старше 20, но моложе 40 лет; 4. —дистрофическая миотония с поздним дебютом(DM with late onset) — у индивидуумов старше 40 лет и с более легким течением. ГЕНЕТИКА При DM1 мутация заключается в экспансии тринуклеотидного повтора ЦТГ (Цитозин-Тимин-Гуадинин) (CTG) в протеинкиназном гене 19-й хромосомы (19q13.3). В норме число CTG-повторов варьирует от 5 до 37, а при DM1 увеличивается от 38 до 2000 и более; наиболее тяжелое течение, ранний дебют и серьезный прогноз заболевания отмечаются у индивидуумов с числом тринуклеотидных повторов, равным 3000 и более. Продуктом гена является белок миотонинпротеинкиназа ( dystrophy myotonic protein kinase — DMPK) . Этот белок является тканеспецифичным. В настоящее время выделяют 6 подтипов DMPK, которые локализуются в различных тканях: в скелетной и гладкой мышечной ткани, в миокарде (волокна Пуркинье, вставочные диски кардиомиоцитов), в центральной нервной системе (на апикальной мембране эпендимы, plexus choroideus, в синапсах мозжечка, гиппокампе, продолговатом и среднем мозге), фибробластах, лимфоцитах , что обусловливает вариабельность картины заболевания. Мутация DMPK приводит к нарушению гомеостаза ионов Ca++, а также процессов возбуждения и сокращения мышц, сердечной проводимости. Рождение ребенка с конгенитальной формой DM1 часто является большой неожиданностью для семьи: во-первых, неожиданное сообщение врача (невролога, неонатолога) о рождении больного ребенка с наследственной патологией в семье, где родители считали себя здоровыми людьми, и разъяснение родственникам сути заболевания; во-вторых, неожиданность понимания того, что чаще всего больна и мать ребенка. Иногда диагноз устанавливается после смерти младенца или рождения мертвого ребенка. Эта реальная ситуация объясняется митохондриальным типом наследования DM1, не всегда подчиняющимся законам Менделя. Индивидуумы с умеренным фенотипом DM1 часто не осознают наличия у себя нервно-мышечного заболевания, которое, как правило, активно диагностируется только при выявлении в семье пациента с более тяжелым течением болезни. Это часто происходит, когда мать с асимптомным течением DM1, имеющая размер тринуклеотидного повтора CTG < 100, рождает младенца с врожденной (конгенитальной) DM1 с длиной CTG-повтора в тысячи. |