Значение биохимии в подготовке врача. Биологическая химия
 Скачать 8.33 Mb. Скачать 8.33 Mb.
|

5%) выделяется с мочой.Б. Изменения метаболизма при сазарном диабете При сахарном диабете, как правило, соотношение инсулин/глюкагон снижено. При этом ослабевает стимуляция процессов депонирования гликогена и жиров, и усиливается мобилизация запасов энергоносителей. Печень, мышцы и жировая ткань даже после приёма пищи функционируют в режиме постабсорбтивного состояния. 1. Симптомы сахарного диабета Для всех форм диабета характерно повышение концентрации глюкозы в крови - гипергликемия.После приёма пищи концентрация глюкозы может достигать 300-500 мг/дл и сохраняется на высоком уровне в постабсорбтивном периоде, т.е. снижается толерантность к глюкозе. Снижение толерантности к глюкозе наблюдают в случаях скрытой (латентной) формы 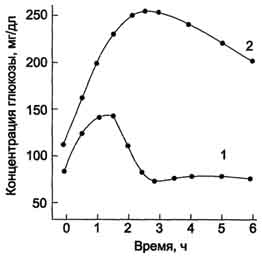 Рис. 11-30. Изменение толерантности к глюкозе у больных скрытой формой сахарного диабета. Определение толерантности к глюкозе используют для диагностики сахарного диабета. Обследуемый принимает раствор глюкозы из расчёта 1 г на 1 кг массы тела (сахарная нагрузка). Концентрацию глюкозы в крови измеряют в течение 2-3 ч с интервалами в 30 мин. 1 - у здорового человека; 2 - у больного сахарным диабетом. сахарного диабета. В этих случаях у людей отсутствуют жалобы и клинические симптомы, характерные для сахарного диабета, а концентрация глюкозы в крови натощак соответствует норме. Однако использование провокационных проб (например, сахарной нагрузки) выявляет снижение толерантности к глюкозе (рис. 11-30). Повышение концентрации глюкозы в плазме крови обусловлено снижением скорости использования глюкозы тканями вследствие недостатка инсулина или снижения биологического действия инсулина в тканях-мишенях. При дефиците инсулина уменьшается количество белков-переносчиков глюкозы (ГЛЮТ-4) на мембранах инсулинзависимых клеток (жировой ткани и мышц). В мышцах и печени глюкоза не депонируется в виде гликогена, в жировой ткани уменьшается скорость синтеза и депонирования жиров. Кроме того, при снижении инсулинглюкагонового индекса активируется глюконеогенез из аминокислот, глицерола и лактата. Повышение концентрации глюкозы в крови при сахарном диабете превышает концентрационный почечный порог, что становится причиной выделения глюкозы с мочой (глюкозурия). В норме проксимальные канальцы почек реабсорбируют всю фильтрующуюся в клубочках глюкозу, если её уровень не превышает 8,9 ммоль/л (160 мг/дл). К характерным признакам сахарного диабета относят также повышение концентрации в крови кетоновых тел - кетонемия.При низком соотношении инсулин/глюкагон жиры не депонируются, а ускоряется их катаболизм, так как гормончувствительная липаза в жировой ткани находится в фосфорилированной активной форме. Концентрация неэтерифицирован-ных жирных кислот в крови повышается. Печень захватывает жирные кислоты, окисляет их до ацетил-КоА, который, в свою очередь, превращается в β-гидроксимасляную и ацетоуксусную кислоты. В тканях ацетоацетат частично декарбоксилируется до ацетона, запах которого исходит от больных сахарным диабетом и ощущается даже на расстоянии. Увеличение концентрации кетоновых тел в крови (выше 20 мг/дл, иногда до 100 мг/дл) приводит к кетонурии. Накопление кетоновых тел снижает буферную ёмкость крови и вызывает ацидоз. 593 Ещё один характерный признак сахарного диабета - повышенный уровень в крови ли-попротеинов (в основном, ЛПОНП) - гипер-липопротеинемия. Пищевые жиры не депонируются в жировой ткани вследствие ослабления процессов запасания, а поступают в печень, где частично превращаются в триацилглицеролы, которые транспортируются из печени в составе ЛПОНП. При сахарном диабете дефицит инсулина приводит к снижению скорости синтеза белков в организме и усилению распада белков. Это вызывает повышение концентрации аминокислот в крови. Аминокислоты поступают в печень и дезаминируются. Безазотистые остатки гликогенных аминокислот включаются в глюконеогенез, что ещё более усиливает гипергликемию. Образующийся при этом аммиак вступает в орнитиновый цикл, что приводит к увеличению концентрации мочевины в крови и, соответственно, в моче - азотемия и азотурия. Высокие концентрации глюкозы, кетоновых тел, мочевины требуют усиленной экскреции их из организма. Поскольку концентрационная способность почек ограничена, резко увеличивается выделение большого количества воды, в результате чего может наступить обезвоживание организма. Выделение мочи у больных возрастает в несколько раз и в некоторых случаях достигает 8-9 л в сутки, но чаще не превышает 3-4 л - полиурия.Потеря воды вызывает постоянную жажду - полидипсия. 2. Острые осложнения сахарного диабета. Механизмы развития диабетической комы Нарушения обмена углеводов, жиров и белков при сахарном диабете могут приводить к развитию коматозных состояний (острые осложнения). Диабетическая кома проявляется в резком нарушении всех функций организма с потерей сознания. Основные предшественники диабетической комы - ацидоз и дегидратация тканей (рис. 11-31). Параллельно кетоацидозу при декомпенсации диабета развивается нарушение водно-электролитного обмена. В его основе лежит гипергликемия, сопровождающаяся повышением осмотического давления в сосудистом русле. Для сохранения осмолярности начинается компенсаторное перемещение жидкости из клеток и внеклеточного пространства в сосудистое русло. Это ведёт к потере тканями воды и электролитов, прежде всего ионов Na+, K+, С1-, НСО3. В результате развиваются тяжёлая клеточная дегидратация и дефицит внутриклеточных ионов (прежде всего К+), затем возникает общая дегидратация. Это приводит к снижению периферического кровообращения, уменьшению мозгового и почечного кровотока и гипоксии. Диабетическая кома развивается медленно, в течение нескольких дней, но иногда может возникнуть и в течение нескольких часов. Первыми признаками могут быть тошнота, рвота, заторможенность. АД у больных снижено. Коматозные состояния при сахарном диабете могут проявляться в трёх основных формах: кетоацидотической, гиперосмолярной и лакто-ацидотической. Для кетоацидотической комы характерны выраженный дефицит инсулина, кетоацидоз, полиурия, полидипсия. Гипергликемия (20-30 ммоль/л), обусловленная инсулиновой недостаточностью, сопровождается большими потерями жидкости и электролитов, дегидратацией и гиперосмоляльностью плазмы. Общая концентрация кетоновых тел достигает 100 мг/дл и выше. При гиперосмолярной коме наблюдают чрезвычайно высокие уровни глюкозы в плазме крови, полиурию, полидипсию, всегда проявляется тяжёлая дегидратация. Предполагают, что у большинства больных гипергликемия обусловлена сопутствующим нарушением функции почек. Кетоновые тела в сыворотке крови обычно не определяются. При лактоацидотической коме преобладают гипотония, снижение периферического кровообращения, гипоксия тканей, приводящая к смещению метаболизма в сторону анаэробного гликолиза, что обусловливает повышение концентрации молочной кислоты в крови (лакто-ацидоз). Разные варианты диабетической комы в чистом виде практически не встречаются. Их возникновение может быть обусловлено разными факторами, например инфекционными заболеваниями, травмами, хирургическими вмешательствами, токсическими соединениями и др. 3. Поздние осложнения сахарного диабета Главная причина поздних осложнений сахарного диабета - гипергликемия. Гипергликемия приводит к повреждению кровеносных сосудов 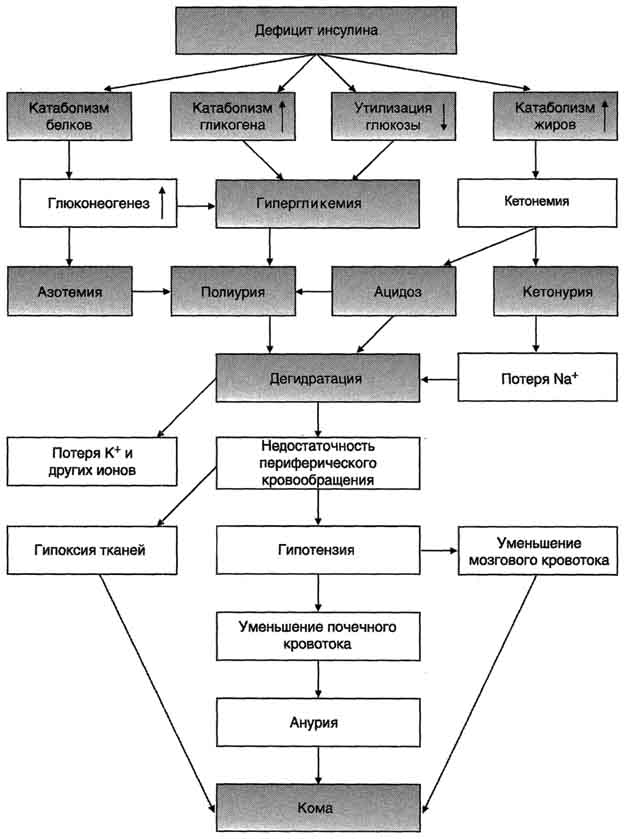 Рис. 11-31. Изменение метаболизма при сахарном диабете и причины диабетической комы. и нарушению функций различных тканей и органов. Одним из основных механизмов повреждения тканей при сахарном диабете являетсягликозилирование белков,приводящее к изменению их конформации и функций. Некоторые белки в норме содержат углеводные компоненты, причём образование таких гликопротеинов протекает ферментативно (например, образование гликопротеиновых гормонов аденогипофиза). Однако в организме человека может происходить и неферментативное взаимодействие глюкозы со свободными аминогруппами белков - неферментативное гликозилирование белков. В тканях здоровых людей эта реакция протекает медленно. При гипергликемии процесс гликозилирования ускоряется. Степень гликозилирования белков зависит от скорости их обновления. В медленно обменивающихся белках накапливается больше изменений. К одним из первых признаков сахарного диабета относят увеличение в 2-3 раза количества гликозилированного гемоглобина (норма НbА1С5,8-7,2%). Другим примером медленно обменивающихся белков служат кристаллины - белки хрусталика. При гликозилировании кристаллины образуют многомолекулярные агрегаты, увеличивающие преломляющую способность хрусталика. Прозрачность хрусталика уменьшается, возникает его помутнение, или катаракта. К медленно обменивающимся белкам относятся белки межклеточного матрикса, базальных мембран. Утолщение базальных мембран, одно из характерных осложнений сахарного диабета, приводит к развитию диабетических ангиопатий. Причиной многих поздних осложнений сахарного диабета также служит повышение скорости превращения глюкозы в сорбитол(см. раздел 7).
Обычно диагноз сахарного диабета можно поставить на основе классических симптомов сахарного диабета - гипергликемии, полиурии, полидипсии, полифагии, ощущения сухости во рту. Важнейшие биохимические признаки ИЗСД выявляют на основе:
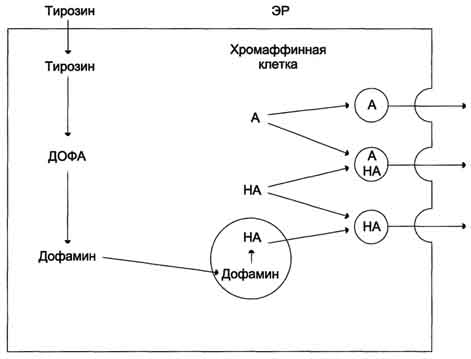
Поскольку ИНСД развивается значительно медленнее, классические клинические симптомы, гипергликемию и дефицит инсулина диагностируют позднее, часто в сочетании с симптомами поздних осложнений сахарного диабета. Сахарный диабет у детей развивается относительно остро, приобретая тяжелое, прогрессирующее течение. Это обусловлено лабильностью нейроэндокринной регуляции обмена, интенсивным ростом организма и высоким уровнем метаболических процессов. В начале заболевания у детей раннего возраста обнаруживаются значительные колебания уровня сахара в крови в течение суток. Склонность детей к кетозу объясняет высокую частоту кетонемии и быстроту развития диабетической комы. Одним из тяжелых проявлений сахарного диабета в детском возрасте является синдром Мориака, который характеризуется значительной задержкой роста, гипогенитализмом, увеличением размеров печени, кетозом, гиперлипидемией и ожирением. При проведении инсулинотерапии у детей чаще, чем у взрослых, может развиться гипогликемическая кома (неустойчивость обмена, анорексия и недостаточное поступление пищи после введения инсулина). Диабетические поражения сосудов, гломерулосклероз с почечной недостаточностью (болезнь Ким-мельстила - Уилсона), ретинопатии и катаракта относятся к поздним последствиям сахарного диабета и у детей наблюдаются редко. 89. Гормоны щитовидной железы. Регуляция синтеза и секреции йодтиронинов и их влияние на метаболизм и функции организма. Изменение метаболизма при гипо- и гипертиреозе. Причины и проявления эндемического зоба. В щитовидной железе синтезируются гормоны - йодированные производные тирозина. Они объединены общим названием йодтирони-ны. К ним относят 3,5,3'-трийодтиронин (трийодтиронин, Т3) и 3,5,3',5'-тетрайодтиронин (Т4), или тироксин. Йодтиронины участвуют в регуляции многих процессов метаболизма, развития, клеточной дифференцировки, в регуляции экспрессии генов. Заболевания, возникающие в результате нарушений синтеза, секреции и функций йодти-ронинов, - наиболее распространённые заболевания эндокринной системы. 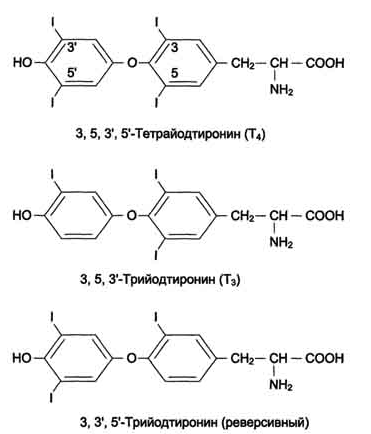 Биосинтез йодтиронинов. Йодтиронины синтезируются в составе белка тиреоглобулина (Тг) в фолликулах, которые представляют собой морфологическую и функциональную единицу щитовидной железы. Тиреоглобулин- гликопротеин с молекулярной массой 660 кД, содержащий 115 остатков тирозина. 8-10% массы тиреоглобулина представлено углеводами. Содержание йодида в организме составляет 0,2-1%. Синтезируется на рибосомах шероховатого ЭР в виде претиреоглобулина, затем переносится в цистерны ЭР, где происходит формирование вторичной и третичной структуры, включая процессы гликозилирования. Из цистерн ЭР Тиреоглобулин поступает в аппарат Гольджи, включается в состав секреторных гранул и секретируется во внеклеточный коллоид, где происходит йодирование остатков тирозина и образование йодтиронинов. Йодирование тиреоглобулина и образование йодтиронинов осуществляется в несколько этапов: 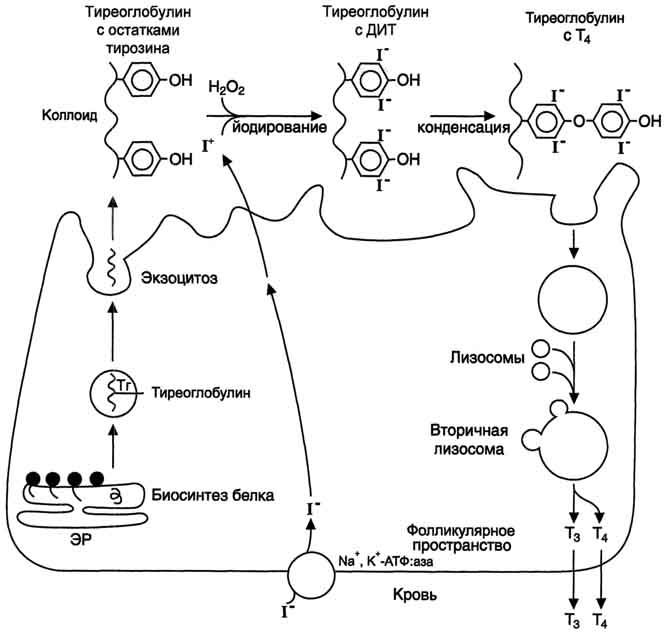 Схема синтеза йодтиронинов. Тиреоглобулин синтезируется на рибосомах, далее поступает в аппарат Гольджи, а затем во внеклеточный коллоид, где он хранится и где происходит йодирование остатков тирозина. Образование йодтиронинов происходит в несколько этапов: транспорт йода в клетки щитовидной железы; окисление йода; йодирование остатков тирозина; образование йодтиронинов; транспорт йодтиронинов в кровь. ЭР - эндоплазматический ретикулум; ДИТ - дийодтиронин; Тг - Тиреоглобулин; Т3 - трийодтиронин, Т4- тироксин. Скорость синтеза и секреции йодтиронинов регулируются гипоталамо-гипофизарной системой по механизму обратной связи. Стимулом для повышения секреции тирео-либерина и тиреотропина служит снижение концентрации йодтиронинов в крови. Регуляция синтеза и секреции одтиронинов. 1 - тиреолиберин стимулирует освобождение ТТГ; 2 - ТТГ стимулирует синтез и секрецию йодтиронинов; 3, 4 - йодтиронины тормозят синтез и секрецию ТТГ и тиреолиберина. При физиологической концентрации йодтиронинов их действие проявляется в ускорении белкового синтеза, стимуляции процессов роста и клеточной дифференцировки. В этом отношении йодтиронины - синергисты гормона роста. Кроме того, Т3 ускоряет транскрипцию гена гормона роста. У животных при дефиците Т3 клетки гипофиза теряют способность к синтезу гормона роста. 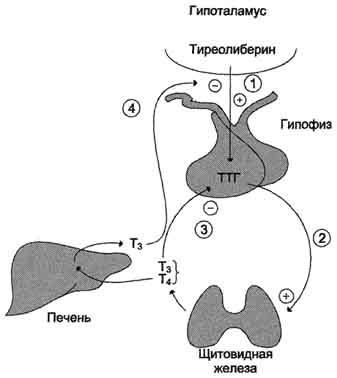 Очень высокие концентрации Т3 тормозят синтез белков и стимулируют катаболические процессы, показателем чего служит отрицательный азотистый баланс. В печени йодтиронины ускоряют гликолиз, синтез холестерола и синтез жёлчных кислот. В печени и жировой ткани Т3 повышает чувствительность клеток к действию адреналина и косвенно стимулирует липолиз в жировой ткани и мобилизацию гликогена в печени. В физ.концентрациях Т3увеличивает в мышцах потребление глюкозы, стимулирует синтез белков и увеличение мышечной массы, повышает чувствительность мышечных клеток к действию адреналина. Йодтиронины также участвуют в формировании ответной реакции на охлаждение увеличением теплопродукции, повышая чувствительность симпатической нервной системы к норадреналину и стимулируя секрецию норадреналина. Гипотиреоз у новорождённыхприводит к развитию кретинизма, который проявляется множественными врождёнными нарушениями и тяжёлой необратимой задержкой умственного развития. Гипотиреозразвивается вследствие недостаточности йодтиронинов. Обычно гипотиреоз связан с недостаточностью функции щитовидной железы, но может возникать и при заболеваниях гипофиза и гипоталамуса. Гипотиреоз может быть также результатом недостаточного поступления йода в организм -эндемический зоб.Эндемический зоб (нетоксический зоб) часто встречается у людей, живущих в районах, где содержание йода в воде и почве недостаточно. Если поступление йода в организм снижается (ниже 100 мкг/сут), то уменьшается продукция йодтиронинов, что приводит к усилению секреции ТТГ (из-за ослабления действия йодтиронинов на гипофиз по механизму отрицательной обратной связи), под влиянием которого происходит компенсаторное увеличение размеров щитовидной железы (гиперплазия), но продукция йодтиронинов при этом не увеличивается. Гипертиреоз возникает вследствие повышенной продукции йодтиронинов. Диффузный токсический зоб(базедова болезнь, болезнь Грейвса) - наиболее распространённое заболевание щитовидной железы. При этом заболевании отмечают увеличение размеров щитовидной железы (зоб), повышение концентрации йодтиронинов в 2-5 раз и развитие тиреотоксикоза. Гипертиреоз может возникать в результате различных причин: развитие опухоли, тиреоидит, избыточное поступление йода и йодсодер-жащих препаратов, аутоиммунные реакции. Болезнь Грейвса возникает в результате образования антител к тиреоидным антигенам. Один из них, иммуноглобулин (IgG), имитирует действие тиреотропина, взаимодействуя с рецепторами тиреотропина на мембране клеток щитовидной железы. Это приводит к диффузному разрастанию щитовидной железы и избыточной неконтролируемой продукции Т3 и Т4, поскольку образование IgG не регулируется по механизму обратной связи. Уровень ТТГ при этом заболевании снижен вследствие подавления функции гипофиза высокими концентрациями йодтиронинов. 90. Гормоны коры надпочечников (кортикостероиды). Их влияние на метаболизм клетки. Изменения метаболизма при гипо- и гиперфункции коры надпочечников. В коре надпочечников синтезируется более 40 различных стероидов, различающихся по структуре и биологической активности. Биологически активные кортикостероиды объединяют в 3 основные класса в зависимости от их преобладающего действия. Глюкокортикоиды,С21-стероиды, играют важную роль в адаптации к стрессу. Они оказывают разнообразные эффекты, но наиболее важный - стимуляция глюконеогенеза. Основной глюкокортикоид человека - кортизол. Минералокортикоиды,С21-стероиды, необходимы для поддержания уровня Na+ и К+. Самый активный гормон этого класса - альдостерон. Андрогены - С19-стероиды. В коре надпочечников образуются предшественники андрогенов, из которых наиболее активный - дегидроэпиандростерон (ДЭА) и слабый - андростендион. Самый мощный андроген надпочечников тестостерон синтезируется в надпочечниках в небольшом количестве. Эти стероиды превращаются в более активные андрогены вне надпочечников. Тестостерон в незначительных количествах может превращаться в надпочечниках в эстрадиол. Но в норме продукция этих гормонов надпочечниками не играет существенной роли. Строение и основные этапы синтеза кортикостероидов. 1 - превращение холестерола в прегненолон (гидроксилаза, отщепляющая боковую цепь); 2 - образование прогестерона (3-β-гидроксистероиддегидрогеназа);3,4,5 - реакции синтеза кортизола (3 - 17-гидроксилаза, 4 - 21-гидроксилаза, 5 - 11-гидроксилаза); 6, 7, 8 - путь синтеза альдостерона (6 - 21-гидроксилаза, 7 - 11-гидроксилаза, 8 - 18-гидроксилаза, 18-гидроксидегидрогеназа); 9,10,11 - путь синтеза тестостерона (9 - 17-гидроксилаза, 10 - 17,20-лиаза, 11 - дегидрогеназа). 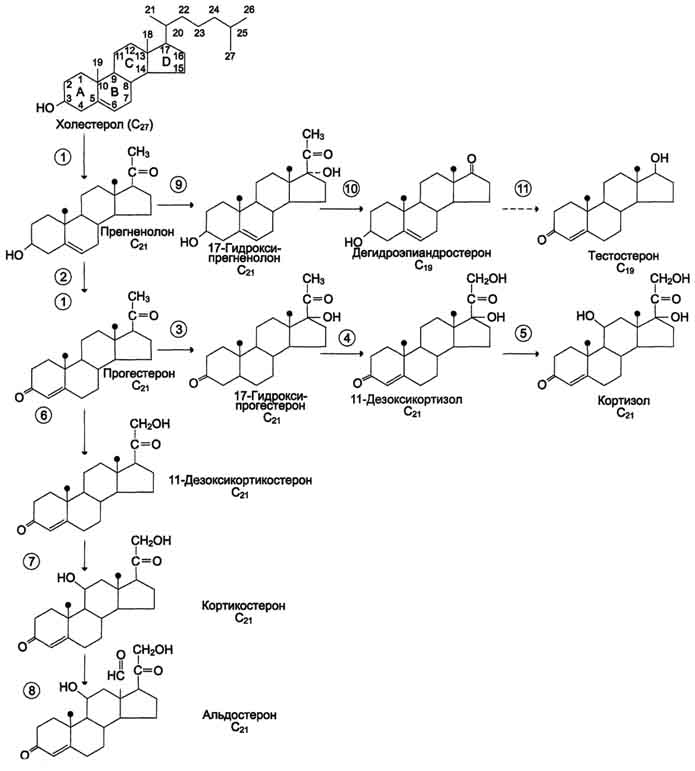 Биологические функции кортикостероидовотличаются широким спектром влияний на процессы метаболизма и подробно рассматриваются в соответствующих разделах. Важнейший фактор в механизме действия кортикостеровдов - взаимодействие их со специфическими рецепторами, расположенными в цитозоле клетки или в ядре. Регуляция внутриклеточных процессов под влиянием кортико-стероидных гормонов проявляется в изменении количества белков, обычно ключевых ферментов метаболизма, путём регуляции транскрипции генов в клетках-мишенях. Влияние глюкокортикоидовна промежуточный метаболизм связано с их способностью координированно воздействовать на разные ткани и разные процессы, как анаболические, так и катаболические. Кортизол стимулирует образование глюкозы в печени, усиливая глюконеогенез и одновременно увеличивая скорость освобождения аминокислот - субстратов глюконеогенеза из периферических тканей. В печени кортизол индуцирует синтез ферментов катаболизма аминокислот (аланинаминотрансферазы, трипто-фанпирролазы и тирозинаминотрансферазы и ключевого фермента глюконеогенеза - фосфоенолпируваткарбоксикиназы). Кроме того, кортизол стимулирует синтез гликогена в печени и тормозит потребление глюкозы периферическими тканями. Это действие кортизола проявляется в основном при голодании и недостаточности инсулина. У здоровых людей эти эффекты кортизола уравновешиваются инсулином. Избыточное количество кортизола стимулирует липолиз в конечностях и липогенез в других частях тела (лицо и туловище). Кроме того, глюкокортикоиды усиливают липолитическое действие катехоламинов и гормона роста. Влияние глюкокортикоидов на обмен белков и нуклеиновых кислот проявляется двояко: в печени кортизол в основном оказывает анаболический эффект (стимулирует синтез белков и нуклеиновых кислот). В мышцах, лимфоидной и жировой ткани, коже и костях кортизол тормозит синтез белков, РНК и ДНК и стимулирует распад РНК и белков. При высокой концентрации глюкокортикоиды подавляют иммунные реакции, вызывая гибель лимфоцитов и инволюцию лимфоидной ткани; подавляют воспалительную реакцию, снижая число циркулирующих лейкоцитов, а также индуцируя синтез липокортинов, которые ингибируют фосфолипазу А2, снижая таким образом синтез медиаторов воспаления - простагландинов и лейкотриенов. Высокая концентрация глюкокортикоидов вызывает торможение роста и деления фибро-бластов, а также синтез коллагена и фибронектина. Для гиперсекреции глюкокортикоидов типичны истончение кожи, плохое заживление ран, мышечная слабость и атрофия мышц. Глюкокортикоиды участвуют в физиологическом ответе на стресс, связанный с травмой, инфекцией или хирургическим вмешательством. В этом ответе в первую очередь участвуют кате-холамины, но во многих случаях для проявления их максимальной активности необходимо участие глюкокортикоидов. Минералокортикоидыстимулируют реабсорбцию Na+ в дистальных извитых канальцах и собирательных трубочках почек. Кроме того, они способствуют секреции К+, NH4+ в почках, а также в других эпителиальных тканях: потовых железах, слизистой оболочке кишечника и слюнных железах. В организме человека альдостерон - наиболее активный минералокортикоид. Изменения метаболизма при гипо- и гиперфункции коры надпочечников Заболевания коры надпочечников могут проявиться симптомами как гипо-, так и гиперпродукции гормонов. Большинство клинических проявлений надпочечниковой недостаточности обусловлено дефицитом глюкокортикоидов и минералокортикоидов. Острая надпочечниковая недостаточность представляет большую угрозу для жизни, так как сопровождается декомпенсацией всех видов обмена и процессов адаптации. Она проявляется сосудистым коллапсом, резкой адинамией, потерей сознания. Такое состояние возникает вследствие нарушения обмена электролитов, которое приводит к потере ионов Na+ и Сl- с мочой, обезвоживанию за счёт потери внеклеточной жидкости, повышению уровня К+ в сыворотке крови, в межклеточной жидкости и клетках, в результате чего может нарушаться сократительная способность миокарда. Изменение углеводного обмена проявляется в снижении уровня сахара в крови, уменьшении запаса гликогена в печени и скелетных мышцах. Острая недостаточность функции коры надпочечников может быть следствием декомпенсации хронических заболеваний, а также развивается у больных, лечившихся длительное время глюкокортикоидными препаратами по поводу неэндокринных заболеваний, например инфекционно-аллергических заболеваний. В результате длительного приёма глюкокортикоидов подавляется функция гипоталамо-гипофизарно-надпочечниковой системы и развивается атрофия клеток коры надпочечников. Резкая отмена гормональных препаратов может сопровождаться острой надпочечниковой недостаточностью (так называемый синдром "отмены"). Первичная недостаточность надпочечников (болезнь Аддисона)развивается в результате поражения коры надпочечников туберкулёзным или аутоиммунным процессом. Основные клинические проявления выражаются в снижении массы тела, общей слабости, снижении аппетита, тошноте, рвоте, снижении АД и типичной для первичной надпочечниковой недостаточности гиперпигментацйи кожи ("бронзовая болезнь") . Причина гиперпигментации - повышение продукции ПОМК - предшественника АКТГ и меланоцитстимулирующего гормона. Вторичная недостаточность надпочечниковможет развиться при дефиците АКТГ, что, в свою очередь, может быть следствием опухоли или инфекционного поражения гипофиза. При вторичной недостаточности надпочечников, в отличие от болезни Аддисона, отсутствует гиперпигментация. При врождённой гиперплазии надпочечниковнарушается синтез кортизола. В 95% случаев при этой патологии обнаруживается дефект 21-гидроксилазы (реже 11-гидроксилазы). Снижение продукции кортизола сопровождается увеличением секреции АКТГ, накоплением промежуточных продуктов синтеза кортикостероидов, в частности, предшественников андрогенов. Избыток андрогенов ведёт к усилению роста тела, раннему половому созреванию у мальчиков и развитию мужских половых признаков у девочек (адреногенитальный синдром). При частичной недостаточности 21-гидроксилазы у женщин может нарушаться менструальный цикл. Гиперпродукция глюкокортикоидов (гиперкортицизм)может быть следствием повышения уровня АКТГ при опухолях гипофиза (болезнь Иценко-Кушинга)и опухолях других клеток (бронхов, тимуса, поджелудочной железы), вырабатывающих кортикотропинподобные вещества, или избыточного синтеза кортизола при гормонально-активных опухолях коры надпочечников (синдром Иценко-Кушинга). При гиперкортицизме наблюдаются гипергликемия и снижение толерантности к глюкозе, обусловленные стимуляцией глюконеогенеза ("стероидный диабет"), усиление катаболизма белков, уменьшение мышечной массы, истончение кожи, остеопороз, инволюция лимфоидной ткани. Характерно своеобразное перераспределение отложений жира ("лунообразное лицо", выступающий живот). Гипернатриемия, гипертензия, гипокалиемия обусловлены некоторой минералокортикоидной активностью кортизола, которая проявляется при его избытке. Для выявления первичной причины гиперкортицизма, помимо определения концентрации АКТГ в плазме крови, используют тесты с применением высоких доз синтетического глюкокортикоида дексаметазона (структурного аналога кортизола). Дексаметазон подавляет секрецию АКТГ по механизму отрицательной обратной связи. Для болезни Иценко-Кушинга характерно снижение концентрации кортизола после применения дексаметазона более чем на 50%. Отсутствие реакции на введение дексаметазона может указывать на наличие опухоли надпочечников или внегипофизарной секреции АКТГ. 91. Гормоны мозгового слоя надпочечников. Секреция катехоламинов. Механизм действия и биологические функции катехоламинов. Патология мозгового вещества надпочечников. Мозговой слой надпочечников - производное нервной ткани. Его можно рассматривать как продолжение симпатической нервной системы, так как преганглионарные волокна чревного нерва оканчиваются на хромаффинных клетках мозгового слоя надпочечников. Своё название эти клетки получили потому, что они содержат гранулы, окрашивающиеся бихроматом калия в красный цвет. Такие клетки находятся также в сердце, печени, почках, половых железах, постганглионарных нейронах симпатической нервной системы и в ЦНС. При стимуляции преганглионарного нейрона хромаффинные клетки продуцируют катехоламины - дофамин, адреналин и норадреналин. У большинства видов животных хромаффинные клетки секретируют в основном адреналин ( |