Зачёт клин патфиз. Patfiz_zachet переделанный. 1. Понятие о клинической патофизиологии, ее задачи и перспективы
 Скачать 5.74 Mb. Скачать 5.74 Mb.
|

|
1.Понятие о клинической патофизиологии, ее задачи и перспективы Клиническая патофизиология- это раздел патофизиологии, изучающий основные закономерности развития ведущих клинических форм патологии у больных людей с помощью безвредных для них методов исследования (физиологических, биохимических, ультразвуковых, иммунологических, КТ и тд.) Задачи клинической патофизиологии: 1. Исследование общих и частных закономерностей развития болезней у больного человека. Установление их связи с реактивностью организма, а также влиянием на их реализацию личности врача и содержания терапии. 2. Изучение патогенеза конкретной болезни у конкретного больного с целью повышения эффективности терапии. 3. Формирование патогенетических принципов терапии, адекватных современному уровню представлений о патогенезе. 2.Перечислить основные типовые расстройства липидного обмена. Привести примеры. 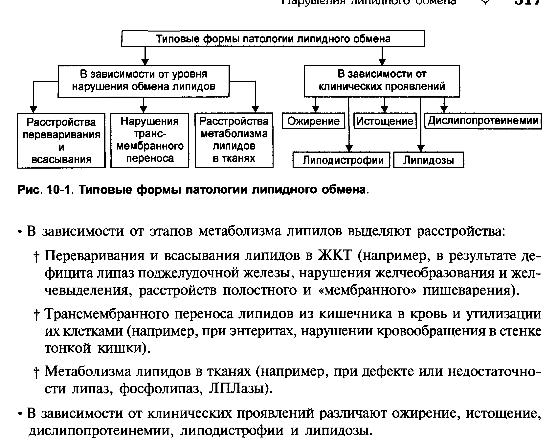 3.«Ожирение» - определение понятия. Классификация.    4.Перечислить основные причины приводящие к развитию ожирения в зависимости от вида ожирения.  5.Ожирение. Нейрогенный механизм возникновения. 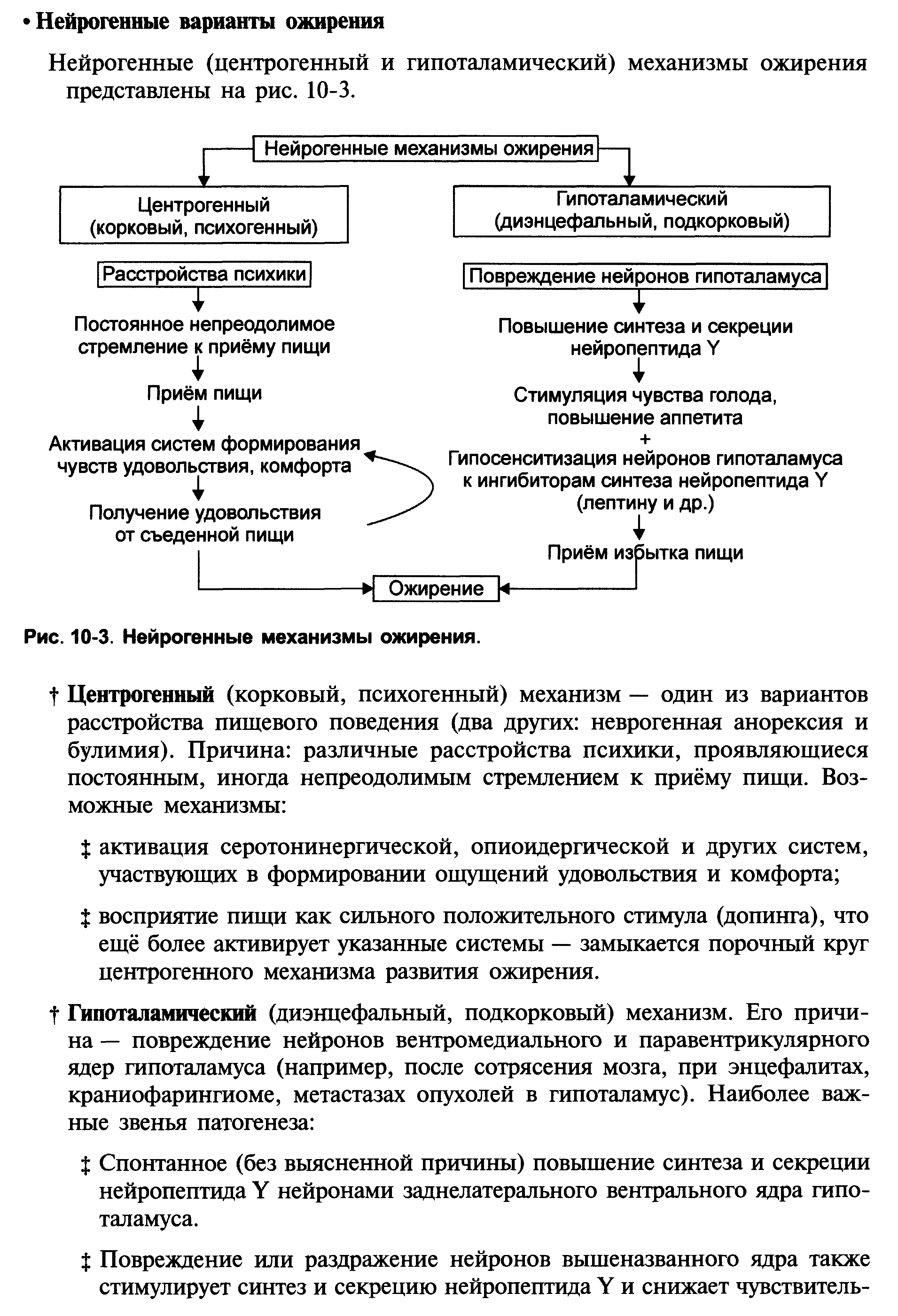  6.Ожирение. Эндокринный механизм возникновения. 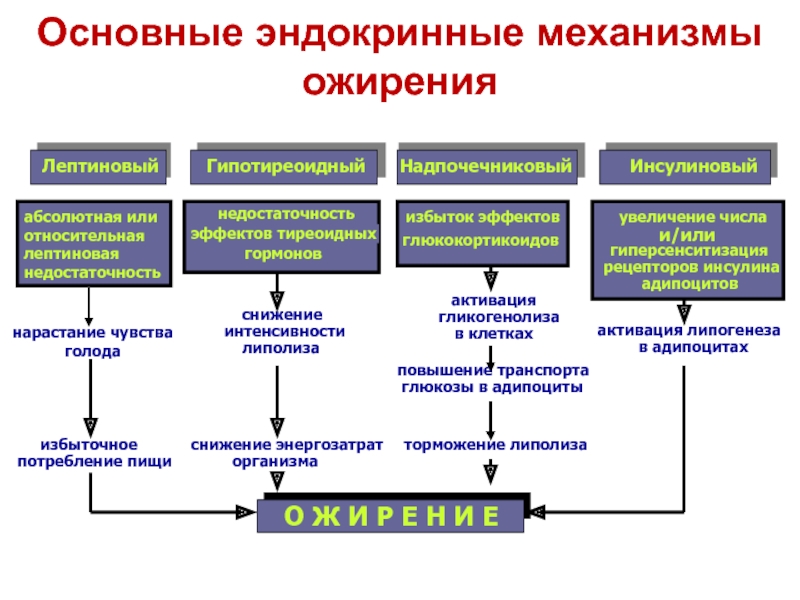 7.Истощениеи кахексия. Определение. Этиология первичного и вторичного вида. Истощение и кахексия — патологическое снижение массы жировой ткани ниже нормы. Одновременно значительно снижается масса мышечной и соединительной ткани. При истощении дефицит жировой ткани может составлять 20– 25% и более (при индексе массы тела ниже 20 кг/м2), а при кахексии — ниже 50%. (причины смотреть в таблице) 8.Истощение и кахексия. Основные звенья патогенеза. 9.Перечислить основные звенья патогенеза первичных эндогенных форм истощения и кахексии 10.Назвать основные звенья патогенеза вторичных эндогенных форм истощения Вторичные эндогенные формы истощения и кахексии являются симптомами других форм патологии: синдромов мальабсорбции, роста новообразований (синтезирующих ФНОа), гипоинсулинизма, гипокортицизма, недостатка эффектов гормонов вилочковой железы. 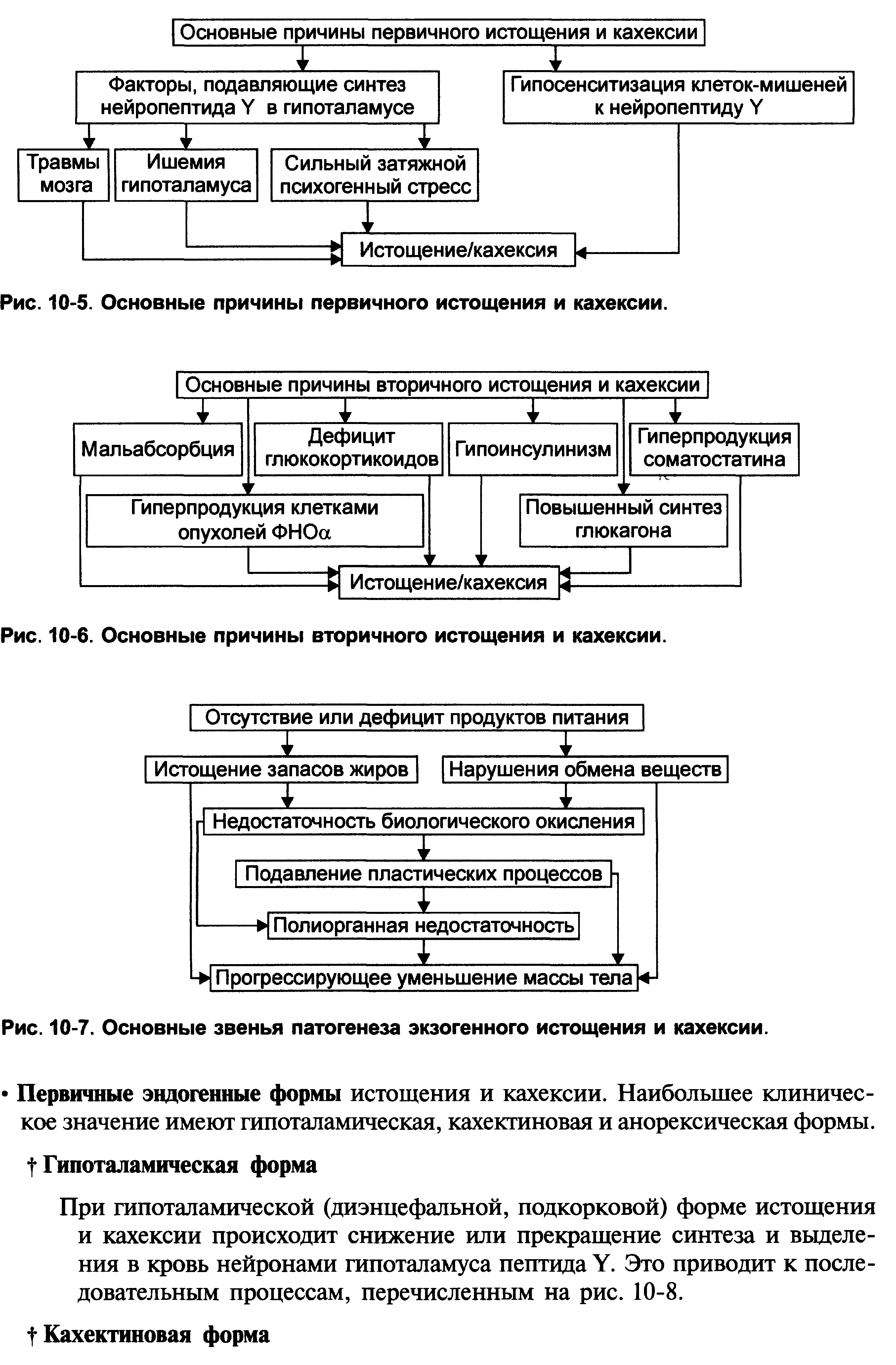 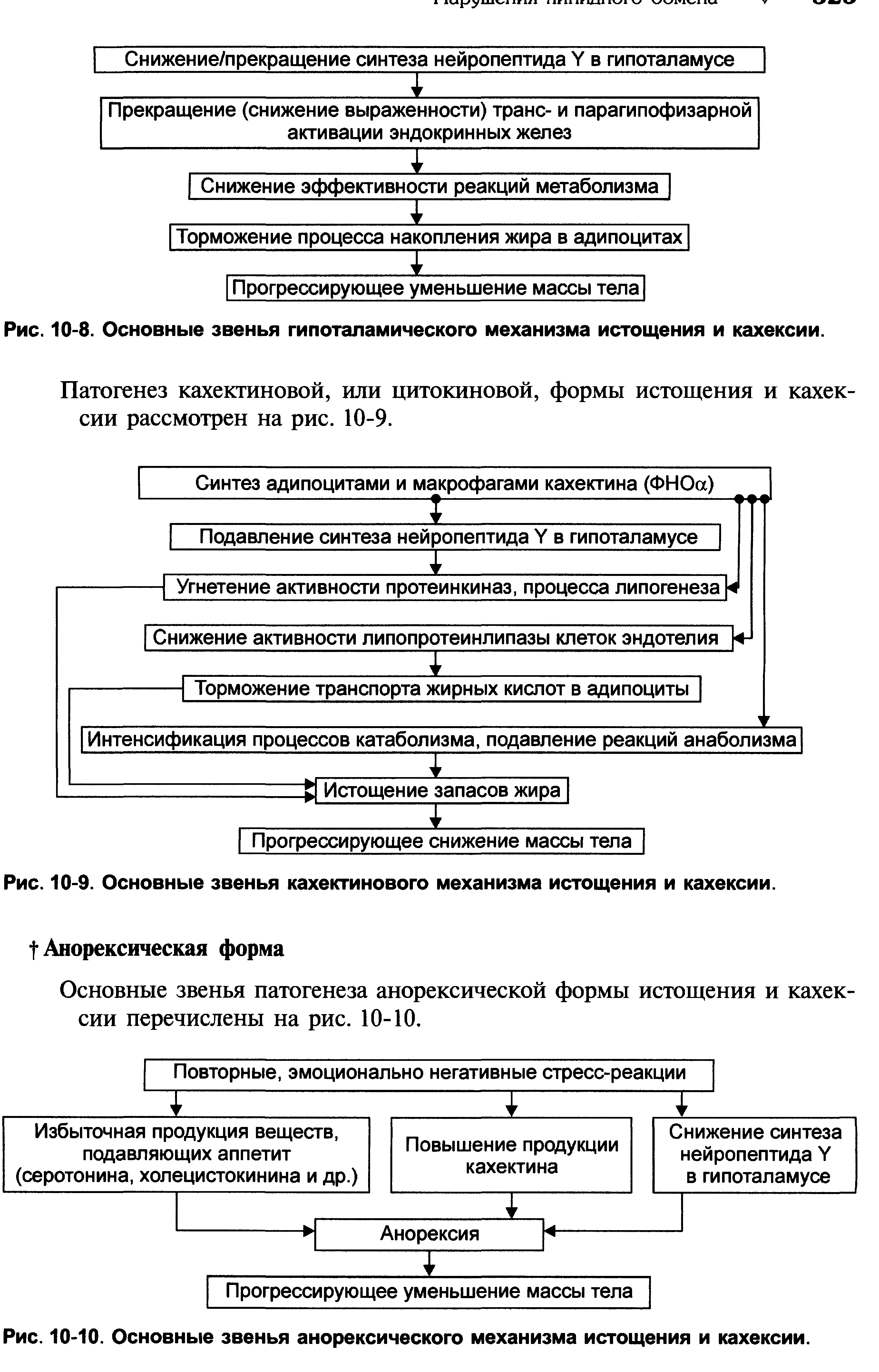 10.Назвать основные звенья патогенеза вторичных эндогенных форм истощения 11.Дислипопротеидемии. Этиология, виды, механизмы развития. 12.Дать характеристику основным классам липопротеидов. Перечислить их функции. Привести расчёт индекса атерогенности.   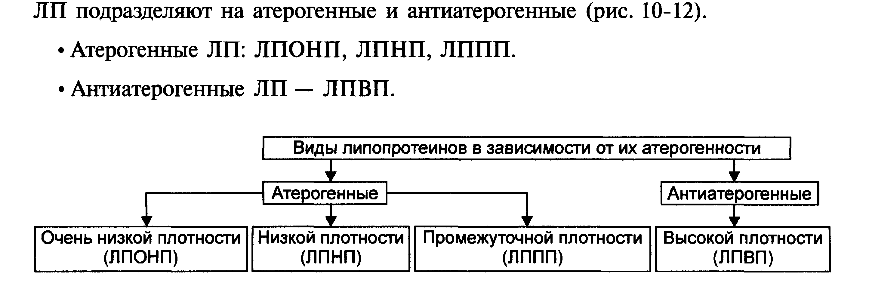   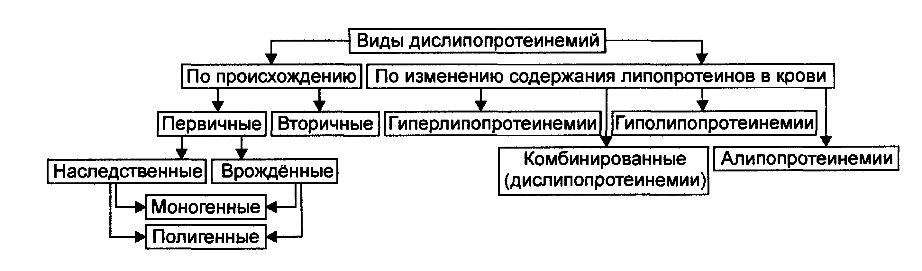  13.«Атеросклероз» - дать определение понятия. Этиология. Перечислить основные факторы риска развития атеросклероза.      14.Атеросклероз – основные механизмы развития по стадиям. Выделяют следующие этапы атерогенеза: – инициация его, – прогрессирование атерогенеза, – формирование атеромы, – образование фиброатеромы, – развитие осложнений атеросклероза.    15.Атеросклероз. Перечислить и пояснить возможные осложнения.  16.Атеросклероз. Основные принципы профилактики и лечения.   17.Этапы образования фибринового сгустка. Внутренний и внешний пути свертывания. Свертывание крови- это каскадно-ферментативный процесс, который протекает в виде последовательных взаимосвязанных стадий и заканчивается образованиемфибринового тромба. Согласно концепции основоположника ферментативной теории гемостаза А.А. Шмидта считается, что свертывание крови включает в себя три фазы: 1) образование активной протромбиназы, 2) образование тромбина, 3) образование фибрина. Фаза I– комплекс последовательных ферментативных реакций, приводящих к образованиюактивной протромбиназы. В зависимости от механизмов, различают два пути образования протромбиназы - внешнийивнутренний.Внешний механизмзапускается при повреждении сосудистой стенки или окружающих тканей. Он завершается образованиемактивной тканевой протромбиназычерез 5-10 секунд. Внутренний механизмзапускается при контакте крови с поврежденным субэндотелием, компонентами соединительной ткани сосудистой стенки или при разрушении форменных элементов крови. Он завершается через 5-7 мин образованиемактивной кровяной протромбиназы Фаза II. Активированная протромбиназа в присутствии ионов кальция за 2-5 сек превращаетпротромбин- неактивный профермент плазмы крови в активныйтромбин. Фаза III.Под влиянием тромбинафибриноген- белок плазмы крови, превращается вфибрин. Процесс превращения фибриногена в фибрин включает в себя три этапа: Из фибриногена образуется фибрин-мономер (фибрин M). Под влиянием ионов кальция фибрин-мономер полимеризуется с образованием растворимого фибрин-полимера (фибрин S, лат. «soluble» – растворимый). Фибринстабилизирующий фактор (ХIII), который активируется в присутствии ионов кальция тромбином, переводит растворимый фибрин в окончательный нерастворимый полимерфибрин (фибрин I, лат. «insoluble» – нерастворимый). Образование нерастворимого полимерфибрина, в нитях которого задерживаются форменные элементы крови, завершает формирование фибринового тромба - кровяного сгустка. Через 30-60 минут после образования тромба начинается его ретракция- уплотнение, которое связано с укорочением тромбоцитарного тромбостенина и сжатием сети фибрина, обеспечивающим более прочную и надежную закупорку поврежденного сосуда. Ретракция заканчивается в течение 2-4 часов после образования кровяного сгустка. Практически одновременно запускается фибринолиз- процесс растворения (лизиса) сгустка крови, который обусловлен ферментативным расщеплением фибрина. В естественных условиях свертывающая и противосвертывающая системы находятся в состоянии динамического равновесия, обеспечивая жидкое состояние крови и, одновременно, готовность к закупорке сосуда при его повреждении. Ускорение свертывания крови называют гиперкоагулемией, а замедление –гипокоагулемией. 18.Причины, механизм и последствия нарушения сосудисто - тромбоцитарного (первичного) гемостаза. Тромбоцитарно-сосудистый (первичный) гемостаз 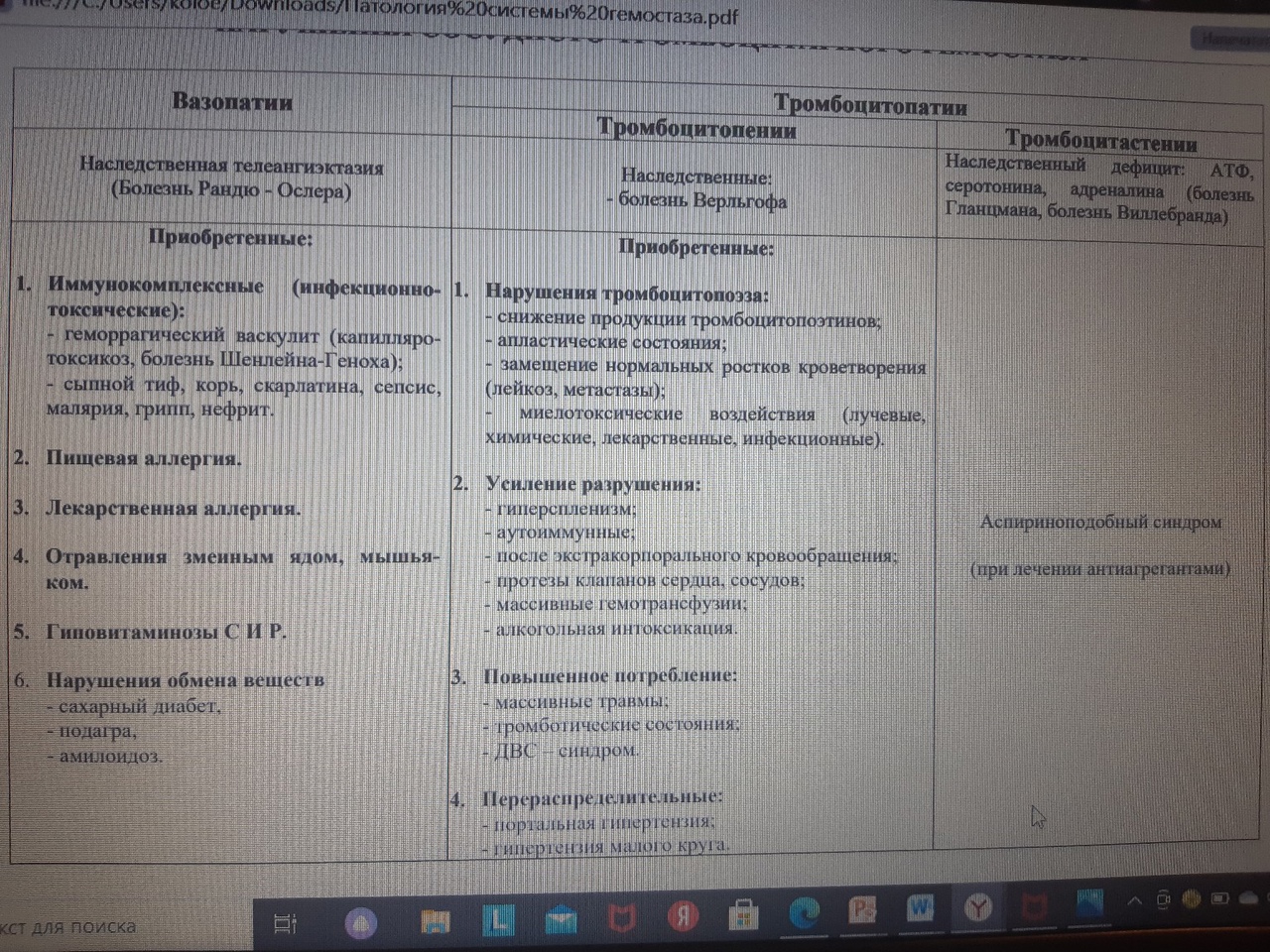 19.Причины, механизм и последствия нарушения коагуляционного (вторичного) гемостаза. Коагуляционный (вторичный) гемостаз  20.Причины и последствия нарушения функции противосвертывающей системы: антикоагулянтов (первичных и вторичных) и системы плазминогена Наследственые и приобретенные нарушения фибринолиза. Приобретенные нарушения фибринолиза встречаются чаще наследственных. Нарушения фибринолиза подразделяются на 2 вида: - гиперфибринолитические состояния – вызваны избыточной его актива- цией или нарушением ингибирования; сопровождаются геморрагическим диатезом; - гипофибринолитические состояния сопровождаются тромбозом. Гиперфибринолитические состояния Избыточная активация фибринолиза может быть вызвана: - повышенным выделением в кровь активаторов плазминогена (ТАП, уро- киназы); - избыточным локальным выделением активатора (при меноррагии, проста- тэктомии); - вторичной активации вследствие тромболитической терапии. Нарушение ингибирования фибринолиза наблюдается при: - заболеваниях печени; - снижении уровня ингибитора активатора плазминогена (ИАП), a2- антиплазмина; - вторично, при образовании комплекса плазмина с антиплазмином и (или) плазмина с ингибитором активатора плазминогена (ИАП). Гипофибринолитические состояния обусловлены: - нарушением активации фибринолиза вследствие неадекватно низкого вы- деления тканевого активатора плазминогена (наследственно обусловленного или приобретенного); - избыточной продукцией ингибиторов активатора плазминогена (наследст- венно обусловлено); - терапией анитифибринолитическими средствами (e-аминокапроновая ки- слота). 21.Этиопатогенетическая классификация нарушений гемостаза. Классификация нарушения гемостаза: - усиление процессов свёртывания белков крови и тромбообразования: гиперкоагуляция и развитие тромботического синдрома; - снижения интенсивности процессов свёртывания белков крови и тромбообразования: гипокоагуляция и развитие геморрагических синдромов; - фазное нарушение состояния системы гемостаза (тромбо-геморрагический синдром). Этиопатогенетическая классификация геморрагических синдромов: 1. Нарушение механизмов первичного гемостаза (тромбоцитарно-сосу- дистой реакции): 1.1. Изменение количества тромбоцитов (тромбоцитопении, тромбоцитемии); 1.2. Нарушение адгезии тромбоцитов (болезнь Виллебранда, тромбоцитопатия Бернара-Сулье); 1.3. Нарушение агрегации (тромбастения Гланцмана, передозировка антиагре- гантов); 1.4. Аномалии сосудистой стенки: врожденные (телеангиэктазии) и приобретенные - инфекционного, иммунного или инфекционно-иммунного генеза (болезнь Шенлейна-Геноха). 2. Нарушение механизмов вторичного (коагуляционного) гемостаза: 2.1. Дефицит прокоагулянтов врожденный (нарушение синтеза антигемофиль- ных глобулинов при гемофилиях; синтез аномального фибриногена); 2.2. Дефицит прокоагулянтов приобретенный (нарушение синтеза прокоагулянтов группы протромбина при патологии печени); 3. Преобладание противосвертываюшей системы: 3.1. Врожденного характера - первичный гиперфибринолиз; 3.2. Приобретенного характера - передозировка антикоагулянтов и/или фибринолитических препаратов. 22.Причины развития тромбофилий. Тромботический синдром или тромбофилия (от гр. thrombos ком, сгусток, phileo люблю) – типовая форма патологии системы гемостаза, характеризующаяся чрезмерной (неадекватной) коагуляцией крови и тромбообразованием, ведущими к ишемии тканей и органов. Причины: - повреждение стенок сосудов и сердца (например, при их механической травме, деструкции атеросклеротической бляшки, васкулитах, ангиопатиях у пациентов с СД); - патология форменных элементов крови (например, тромбоцитопатии, гемолиз эритроцитов, чрезмерное повышение адгезии и агрегации тромбоцитов и эритроцитов); - расстройства системы гемостаза (например, абсолютное или относительное преобладание эффектов прокоагулянтных факторов, либо недостаточность эффектов антикоагулянтных и фибринолитических факторов, Это нередко наблюдается при системном атеросклерозе, сахарном диабете, гипертонической болезни, эндотоксинемиях, шоковых состояниях. 23.Патогенетические факторы тромбообразования. Особенности тромбообразования в артериальных и венозных сосудах. Патогенетические факторы тромбообразования 1. Основной фактор - повреждение сосудистой стенки (структурное или функциональное), приводящее к снижению ее тромборезистентности. При этом ламинарный кровоток, характерный для нормального кровеносного сосуда, сменяется турбулентным. Это способствует задержке форменных элементы сосудистой стенки, их разрушению и выделению тромбопластических веществ. Повреждение эндотелия с обнажением базальной мембраны может осуществляться различными факторами: механическими (катетеризация, травма, острая гипертензия и др.), иммунологическими (наличие специфических антител против клеток эндотелия), химическими, мутагенными,эндотоксинами, вирусами, цитотоксинами (фактор некроза опухолей и др.). 2. Образованию тромбозов способствуют нарушения гемодинамики, в частности, снижение скорости кровотока. Так, очевидно этим объясняется то, что в венах тромбы образуются в 5 раз чаще, чем в артериях; в венах нижних конечностей - в 3 раза чаще, чем в венах верхних конечностей. Тромбообразова- нию может способствовать локальный ангиоспазм, в частности, коронарных сосудов. Замедление кровотока и нарушение микроциркуляции, наряду со сти муляцией синтеза и экспрессией тканевого тромбопластина, лежат в основе тромботических осложнений в очаге воспаления. 3. Тромбообразование может быть обусловлено активацией адгезивно- агрегационной функции тромбоцитов, как первичной (при болезни Вакеза), так и вторичной ( при массивных травмах тканей, хирургических операциях и т.д.). 4. Одним из основных факторов тромбообразования является активация коагуляционного (вторичного) гемостаза. Что наблюдается, в частности, при действии различных повреждающих факторов. 5. К тромбообразованию может привести ослабление противосвертываю- щей системы крови. Так, возможно истощение запасов гепарина, идущего на активацию липопротеиновой липазы при гиперлипидемии; снижение синтеза активаторов плазминогена тканевого (ТПА) и урокиназного типа, а также увеличение продукции ингибиторов этих активаторов. 6. Тромбообразованию способствует нарушение реологических свойств крови, в частности, повышение ее вязкости.Факторами, повышающими вязкость крови, являются следующие: понижение скорости тока крови, повышение содержания в плазме крупнодисперсных белков (глобулинов, фибриногена, тромбина) при снижении содержания альбуминов; повышение содержания ате- рогенных липопротеинов, холестерина, продуктов перекисного окисления липидов; увеличение гематокрита; ацидоз, в частности гиперкапния; понижение деформируемости мембраны эритроцитов; активация агрегации форменных элементов крови (эритроцитов и тромбоцитов). |