УЧЕБНИКгенетика. Генетика изучает процессы преемственности жизни на молекулярном, клеточном, организменном и популяционном уровнях
 Скачать 6.93 Mb. Скачать 6.93 Mb.
|

|
Глава 15 МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ (МГК) Основной целью медико-генетического консультирования является предупреждение рождения ребенка с тяжелыми наследственными заболеваниями, а также консультирование по проблемам планирования семьи. По данным Всемирной организации здравоохранения, в 1990 г. из 10 новорожденных 9 родились с различной степенью генетических патологий и только 1 ребенок из 10 родился «абсолютно» здоровым. И даже если в первое время проявление патологий незаметно, они дадут себя знать подверженностью аллергиям, частыми простудами, сколиозом, астмой или неврозами. А позднее, уже став взрослыми, эти бывшие физиологически незрелые дети будут первыми кандидатами на атеросклероз, диабет, ишемическую болезнь или рак. Первый кабинет по МГК был организован в 1941 г. Дж.Нил в Мичиганском университете (США). В нашей стране в 1932 г. под руководством профессора С.Г. Левита был создан медико-генетический институт, в котором исследовали генетическую природу различных наследственных болезней. Первые медико-генетические кабинеты в России были организованы в Москве и Ленинграде в 1967 г., а с 1970 г. такие кабинеты были организованы при всех республиканских, краевых и областных больницах. В мае 1973 г. кабинет МГК был открыт в Ростове-на-Дону. 15.1. Этапы консультирования Первый этап консультирования начинается с изучения анамнеза болезни, составления генеалогической карты (см.раздел 13.1) и уточнения клинического диагноза, с помощью проведения цитогенетических и биохимических исследований. В некоторых случаях проводят дополнительное обследование родителей, включающее, как правило, дерматоглифическое, цитогенетические исследования (оценка кариотипа родителей: забор периферической крови или соскоб слизистой рта и последующий скрининг хромосом, оценка кариотипа плода с использованием амниоцентеза). Второй этап консультирования — если диагноз уточнен, врач-генетик прогнозирует вероятность рождения больного ребенка. В табл. 15.1: приведены основные варианты генетических задач по оценке риска наследственных болезней. При анализе родословной возможны следующие ситуации, требующие различного подхода: Моногенно наследуемая патология, при которой повторность болезни среди родственников позволяет выяснить тип наследования в данной семье. В этих случаях вычисляют теоретический риск рождения больного ребенка. Полигенно наследуемая патология. Болезнь встречается в различных поколениях у пробанда, но методы теоретического расчета неприменимы, и риск устанавливается на основе эмпирических данных. Такие полигенные болезни, как шизофрения, эпилепсия и др., встречаются часто, о них накоплен достаточный фактический материал, на основе которого рассчитан эмпирический риск, созданы специальные таблицы, где данные риска рассчитаны в зависимости от состояния здоровья родителей уже родившихся детей и других родственников и т.д. (табл. 15.2).    Хромосомные болезни. При аномалиях половых хромосом повторные случаи любой из них в семье исключительно редки. При синдромах XXY и XXX обнаруживается связь с возрастом матери. Наиболее неблагоприятным будет прогноз при транслокациях в тех случаях, когда в гаметах одного из родителей имеется сбалансированная хромосомная мутация (см.раздел 12.2.3). Риск рождения ребенка с синдромом Дауна (трисомия по 21-й хромосоме) увеличивается, если возраст матери превышает 35 лет (табл. 15.3). Больные с синдромом Дауна обычно невысокого роста, отличаются слабоумием и многочисленными физическими пороками (табл.15.4, рис.15.1).  При обнаружении мозаицизма у кого-либо из родителей пробанда риск для сибсов определяется по формуле: 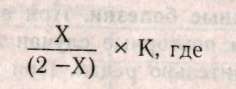 X — доля аномального клеточного клона; К — коэффициент элиминации несбалансированных зигот в эмбриогенезе. Например, при синдроме Дауна К = 0,5.  Случаи единичной патологии, когда анализ родословной не дает указаний на семейный характер болезни. Возможными причинами таких патологий могут быть: генные или хромосомные мутации, возникшие в одной из гамет родителей или на ранних стадиях развития плода; выщепление редкого рецессивного гена вследствие гетерозиготности обоих родителей; сбалансированная транслокация в генотипе одного из родителей; инфекционные болезни у женщины в период беременности (см.раздел 6.4).  Третий этап — официальное заключение с рекомендациями, в т.ч. о степени риска по потомству. Генетический риск оценивают двумя путями: теоретические расчеты, основанные на законах генетики, и на основании эмпирических данных. Степень риска выражается в процентах. В настоящее время считается, что если этот процент составляет 0-10% — это низкий риск, от 11 до 20% — средний и более 21% — высокий. Четвертый этап — совет врача-генетика. Н.П.Бочков (1978) рекомендует проводить заключительный этап через 3-6 месяцев после установления диагноза. Заключение врача-генетика должно быть объективным, так как необоснованный благоприятный совет может обернуться тяжелой психологической травмой после рождения больного ребенка. 15.2. Пренатальная диагностика Пренатальная диагностика плода имеет исключительно важное значение при медико-генетическом консультировании, поскольку позволяет прогнозировать здоровье будущего ребенка в семьях с «отягощенной» наследственностью в I-11 триместре беременности, т.е. когда в случае обнаружения патологии возможно абортирование плода. Наибольшее значение из всех методов прена-тальной диагностики имеет амниоцентез. Амнио-центез — это метод пренатальной диагностики врожденных заболеваний. Его используют при сроках беременности в 15-17 недель в связи с тем, что в это время в амниотической жидкости наблюдается достаточное число живых клеток плода. С помощью хирургического шприца берется проба амниотической жидкости (рис. 15.2). Для определения пола плода достаточно получить 2-5 мл околоплодной жидкости. После центрифугирования осадок наносят отдельными каплями на предметные стекла, высушивают и окрашивают для определения Х- и Y-хроматина. Определение пола важно при заболеваниях, сцепленных с Х-хромосомой. Для определения кариотипа плода необходимо получить 15-20 мл околоплодных вод. Клетки плода из полученной жидкости выращивают в культуральной среде в течение 2 недель. Затем по специальной методике приготавливают пластинки с метафазными хромосомами и проводят цитоге-нетический анализ клеток плода. Если при амнио-цеитезе выявляются серьезные нарушения, такую беременность рекомендуют прекратить. Анализ амниотической жидкости особенно важен при беременностях, сопряженных с высоким риском (особенно в случаях, когда оба родителя — носители одного и того же рецессивного гена). Амниоцентез необходим, если возраст матери превышает 35 лет, когда сильно возрастает вероятность патологий, связанных с нерасхождением хромосом в мейозе. Существуют и другие методы пренатальной диагностики. Ультразвуковое исследование (УЗИ) основано на способности ультразвуковой волны отражаться от поверхности раздела двух сред, отличающихся разной плотностью, что позволяет получить их изображение на экране. Фетоскопия — метод визуального наблюдения плода в полости матки через эластичный зонд, оснащенный оптической системой. 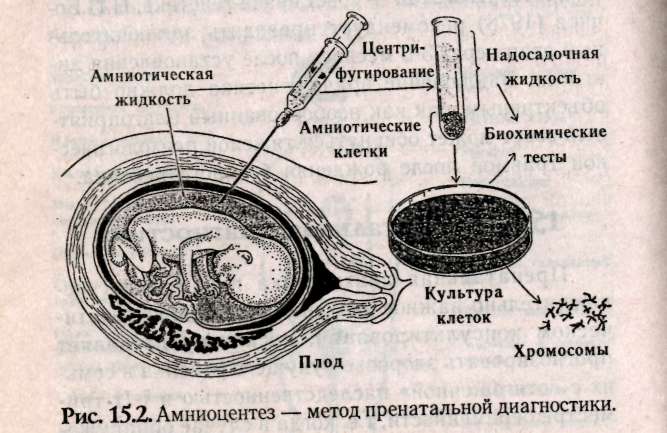 15.3. Планирование семьи Термином «планирование семьи» называются «...те виды деятельности, которые имеют целью помочь отдельным людям или супружеским парам достичь определенных результатов: избежать нежелательной беременности; произвести на свет желанных детей; регулировать интервал между беременностями; контролировать выбор времени деторождения в зависимости от возраста родителей и определять количество детей в семье». ВОЗ (1970) За последние десятилетия исследования в области медицинской генетики выявили еще одно необходимое условие для здоровья детей— планирование семьи. Большинство женщин, которые рожают детей слишком рано, слишком поздно или с небольшим промежутком между родами, рискуют своим здоровьем и здоровьем потомства. Развитие организма женщины протекает так, что она становится фертильной за несколько лет до наступления самого безопасного периода для родов и сохраняет фертильность еще 10-15 лет после окончания этого периода. С биологической точки зрения, наиболее благоприятным возрастом женщин для рождения ребенка считается 20 лет. Период максимальной безопасности длится около 10 лет. Только 10-15% родов падает на возраст до 20 лет. Многие из этих женщин слишком молоды для идеального материнства, как по биологическим, так и по социальным причинам. Они не достигли еще полного физического развития, рискуют родить слабого ребенка, психологические травмы во время беременности серьезнее. После 30 лет возрастает опасность осложнений после родов, возможность рождения недоношенного ребенка или ребенка с недостаточным весом, повышается смертность таких детей. У пожилых матерей чаще появляются дети с врожденными и наследственными пороками. Наиболее показательна в этом отношении частота синдрома Дауна. Вероятность рождения ребенка с синдромом Дауна отчетливо повышается с возрастом матери. Механизм возникновения патологии в данном случае связан с увеличением вероятности нерасхождения хромосом (47, +21) в мейозе при оогенезе. Консультирование по проблемам планирования семьи помогает пациентам выбрать и правильно пользоваться различными способами контрацепции. Роль МГК в настоящее время особенно актуальна в связи с глобальным экологическим кризисом, когда мы живем в среде с повышенной мутагенной активностью, употребляем в пищу не всег- да чистые продукты, дышим, как правило, загрязненным воздухом. Более двух тысяч наследственно обусловленных патологий существует в настоящее время. Под угрозой самое большое богатство — генофонд, хранящий все многообразие наследства, доставшееся от предшествующих поколений, который должен обеспечить потомков наследственностью, необходимой для нормальной жиз ни в будущем. В связи с этим важнейшими задачами на современном этапе являются борьба против возникновения и накопления отрицательных мутаций, снижающих жизнеспособность людей, устранение мутагенов, поиски антимутагенов и главное — всестороннее изучение генетического действия факторов цивилизации и среды на человека. 15.4. Дополнение. Дерматоглифика как метод лабораторно-клинической диагностики Дерматоглифика позволяет диагностировать многие наследственные болезни, среди которых все хромосомные синдромы, большинство пороков развития, многие болезни обмена веществ. И хотя недостаточно разработаны генетические аспекты дерматоглифики, при хромосомных болезнях и врожденных пороках развития дерматоглифика позволяет проводить дифференциальную диагностику с фенокопиями и уточнять клинический диагноз. Кроме того, она позволяет дифференцировать различные нозологические формы. Так было показано, что «взрослая» и «детская» формы диабета представляют собой разные заболевания. Недавно обнаружено, что в ряде случаев изменения дерматоглифики однотипны у пробанда и родителей. Следовательно, можно выявить носительство му-тантных генов по их фенотипическому (дерматог-лифическому) проявлению у здоровых индивидуумов. Дерматоглифический подход относится к экспресс-методам, так как получение и расшифровка занимает не более 20 мин. Этот метод широко используется в работе МГК при диагностике целого ряда патологий. Выявляемость некоторых синдромов при МГК имеет высокую степень достоверности — 90-98%: трисомия-13 (синдром Патау), при пальмос-копии (анализ ладонных узоров) наблюдаются дис-тальные осевые трирадиусы (угол — atd = 180°), радиальные петли на 4-м и 5-м пальцах рук, увеличение удельного веса дуг в узорах пальцев рук и ног, окончание главной линии А у радиального края ладони, наличие дополнительных узоров на гипотенаре, дуговые или Т-образные узоры на поле большого пальца стопы; трисомия-18 (с. Эдвадрса) — дуги не менее, чем на 6 пальцах, а в 80% случаев — на всех пальцах рук и ног. На пятом пальце, а иногда и на всех пальцах отсутствует дистальная сгибательная складка, на поле большого пальца стопы — дуговой узор; трисомия-21 (с. Дауна) — дистальное смещение осевого трирадиуса (L atd = 8Г), ульнар-ная петля на втором пальце и радиальная на четвертом и пятом пальцах, увеличение удельного веса ульнарных петель на пальцах, отсутствие дисталь-ной сгибательной складки на мизинце, учащение узоров на гипотенаре, снижение общего гребневого счета TRC, непрерывистость папиллярных линий; синдром «кошачьего крика» (46,—5р) — увеличение удельного веса завитков и дуг, уменьшение радиальных петель, наличие промежуточно-, го осевого трирадиуса, горизонтальное направление папиллярных линий, уменьшение общего числа гребневого счета TRC; синдром Клайнфельтера (46, XXY) — наиболее часты увеличение удельного веса дуг, снижение гребневого счета, проксимальное смещение осевого трирадиуса, повышение частоты узора на гипотенаре; синдром Шерешевского — Тернера (45, ХО) — отмечаются увеличение удельного веса завитков и уменьшение удельного веса дуг, учащение узора на гипотенаре, вертикальная направленность линий ладоней, снижение частоты узоров на тенаре; трисомия X (47, XXX) — характеризуется увеличением удельного веса завитков и дуг, снижением общего гребневого счета TRC. Изменения дерматоглифики обнаружены при множественных врожденных пороках развития: пороках развития ЦНС, сердечно-сосудистой системы, желудочно-кишечного тракта и др. Олигофрения — имеются четкие половые различия в дерматоглифах: у мужчин — увеличение частоты радикальных петель при уменьшении встречаемых завитков, увеличение TRC; у женщин — уменьшение частоты на гипотенаре и второй межпальцевой подушечки, уменьшение TRC. Шизофрения — отмечается снижение частоты завитков, увеличение дисплазии папиллярных линий, расщелины губы и неба, наличие дистального осевого трирадиуса, мелкобороздчатость. Пороки сердца — при различных типах пороков — повышение удельного веса ульнарных петель, снижение числа дуг и завитков. Дистальное положение осевого трирадиуса, специфика узоров на стопах и ладонях. Глава 16 ПРОБЛЕМЫ РАКА В 1773 г. Лионская академия объявила конкурс на тему «Что такое рак?» Более 200 лет назад писали, что «...рак — это такое заболевание, которое так же трудно определить, как и вылечить», но и в наши дни эта проблема остается актуальной. 1960 г. — создано Международное агентство по изучению рака (МАИР). Основной целью МАИР является обобщение научной информации, получаемой в различных лабораториях мира в области онкологии (науки о раке). Согласно данным МАИР, 80-90% случаев рака вызываются веществами, находящимися в- окружающей человека среде. По данным ВОЗ (Всемирной Организации Здравоохранения), рак является второй из наиболее распространенных причин смертности в мире после сердечно-сосудистых патологий. Рак — это заболевание, связанное с развитием в организме человека злокачественной опухоли. У человека встречаются и доброкачественные опухоли, клетки которой растут, оставаясь в пределах той ткани, где они локализуются. Такие опухоли легко удаляются, не осложняя здоровья человека. Наиболее важные отличия злокачественной опухоли от доброкачественной -- это инвазия и метастазирование. В процессе инвазии опухолевые клетки прорастают в нормальные ткани, нарушают их функционирование и питание, что приводит в конечном итоге к гибели нормальных клеток. Метастазирование — способность злокачественной опухоли образовывать опухолевые узлы в отдаленных частях организма. На определенном этапе развития злокачественных опухолей клетки приобретают способность к амебовидному движению и начинают вторгаться через кровеносную и лимфатическую системы в окружающие ткани и распространяются по всему организму (этап метастазирова-ния). Отличительной чертой злокачественной опухоли является не столько ускоренный рост определенных клеток, сколько нарушение механизмов контроля, не допускающих вторжения клеток в другие ткани и распространения их по организму. Обычно деление здоровых клеток любой ткани контролируется механизмами, заставляющими работать клетку в запрограммированном генотипом режиме и не позволяющими им вторгаться в другие ткани организма. В какой-то момент обычная клетка начинает делиться безудержно, приобретая «бессмертие». Раковые клетки отличаются от нормальных тремя главными свойствами: 1) быстро делятся; 2) сцеплены друг с другом не так прочно, как нормальные; 3) они дедифференцируются и не дифференцируются при последующих делениях и напоминают слабо дифференцированные клетки зародыша. Раковые опухоли в зависимости от локализации в тканях принято разделять на три типа: карциномы (возникают на внутренних оболочках полостных органов и коже); лейкозы (возникают в кроветворных органах — костном мозге, лимфатических узлах, селезенке); саркомы (нарушения клеток соединительной и костной ткани). В 1964 году Дарлингтон попытался обосновать теорию рака соматическими мутациями. Наиболее интересной из разнообразных теорий рака была вирусно-генетическая, предложенная Зилбером в 1968 году. Основные положения этой теории сформулированы в 1975 г. Л.Зилбером, И.Ирлиным и Ф.Киселевым и сводились к следующему: возникновение опухолей вызывается вирусами; механизм вирусного канцерогенеза не является инфекционным, хотя природа онковирусов не отличается от вирусов, вызывающих инфекционные заболевания; вирус является носителем фактора, вызывающего наследственные изменения в клетках; эти изменения нарушают взаимоотношения между клетками и вызывают неконтролируемый рост клеток, приводящий к образованию опухоли. Говоря современным языком, авторы предположили, что ДНК онкогенного вируса при попадании в здоровую клетку встраивается в клеточную ДНК и изменяет ее. В таком виде рекомбинирован-ная ДНК передается следующим клеточным поколениям. Но эта Теория не согласовывалась с тем фактом, что у многих онкогенных вирусов генетическим материалом является РНК, а не ДНК. В 1976 г. Темин и Балтимор обнаружили фермент, названный ревертазой (обратная транскриптаза), который катализирует синтез ДНК (кДНК) по вирусной РНК. В дальнейшем кДНК. при попадании в клетку и вызывает злокачественное перерождение клетки. Это был триумф вирусно-генети-ческой теории. Оставалось идентифицировать онковирусы человека. Но вирусы рака человека так и не удалось обнаружить. Наряду с этим оказалось, что некоторые формы рака имеют не вирусное происхождение. Было показано, что многие химические вещества (канцерогены) способны индуцировать раковую опухоль. Оказалось, что многие мутагены являются сильными канцерогенами. Мутационная теория рака была сформулирована в 1974 году Бернетом: опухоль берет начало от одной исходной соматической клетки; изменения вызываются химическими агентами и вирусами, взаимодействующими с ДНК; мутации могут накапливаться, что приводит клетки к неограниченному размножению; накопление мутаций увеличивается с возрастом человека. В 1975 году Брюс Эймс предложил проверять вещества на мутагенность. Он проверил гипотезу об идентичности понятий «канцерогены» и «мутагены». В 90 случаях из 100 канцерогены оказались сильными мутагенами, но только 13% соединений, не обладающих канцерогенной активностью, оказались мутагенами. Следовательно, события, приводящие в итоге к онкологическим заболеваниям, начинаются с изменений, происходящих в ДНК клеток. К 1978 году формируется генная теория рака, которая преодолела недостатки вирусной и мутационной теорий. В начале 70-х годов Дж.Майкл Бишоп и Гарольд Вармус предположили, что онкогены являются обычными компонентами нормальных кле- ток. В то время многие исследователи считали, что онкогены попадают в организм с вирусами, но Бишоп и Вармус (1976) обнаружили, что онкогены из хромосомной ДНК клетки-хозяина могут включаться в ретровирусный геном (РНК-содержащие вирусы). В' норме эти протоонкогены становятся онкогенами и могут вызывать у человека рак как в случае ретровирусной трансформации, так и в результате воздействия канцерогенов. За это открытие Бишоп и Вармус получили Нобелевскую премию в 1989 г. В 1979 г. Роберт Вайнберг провел серию экспериментов по генетической трансформации, результаты которых показали, что в ДНК человека есть участки — онкогены, активизация которых способствует развитию болезни. У здорового человека онкогены находятся в неактивном состоянии. Вайнберг и его коллеги обнаружили, что протоонкоген (предшественник онкогена) и онкоген отличаются в структурной части белка одним нуклеотидом. В белке, производимом одним из онкогенов, на 12-м месте стоит валин, а у протоонкогена — глицин. К началу 90-х годов было описано около 30 онкогенов: среди них более 20 клеточных онкогенов (с-onc) и еще более 10 проонкогенов несут различные опухолевые вирусы. На рис. 16.1 показана локализация проонкогенов в клетке, а на рис. 16.2 локализация идентифицированных онкогенов в хромосомах человека. В табл. 16.1 описаны патологии, индуцируемые данными онкогенами. Основными принципами генной теории рака в настоящее время можно считать следующие: рак — индуцированное заболевание; ДНК протоонкогенов является мишенью для канцерогенных факторов (мутагенов физической, химической и биологической природы); механизм превращения прото-онкогена в онкоген связан с появлением точковых мутаций в протоонкогене, внутригенных перестройках или с включением в протоонкоген транс-позонов (подвижных элементов ДНК) с последующей амплификацией (образование дополнительных копий) онкогенов; сопутствующие изменения функционирования многих генов являются вторичными факторами канцерогенеза, приводящими к нарушениям сбалансированности нормальной генотипической среды; скрытый период развития опухоли протекает до тех пор, пока действия вторичных факторов канцерогенеза не достигнут критического уровня. Выделены активные онкогены некоторых опухолей человека, экстрагированная ДНК которых переносит способность к неопластическому (злокачественному) росту клеток в культуре ткани. Оказалось, что выделенные из различных опухолей человека онкогены часто оказывались одинаковыми. Предполагается вероятным, что небольшая группа онкогенов участвует в возникновении многих форм рака. На рис.16.3 показана схема возможного общего механизма возникновения спонтанных и индуцированных опухолей. Число соматических клеток в организме отдельного человека в среднем равно 11-14 миллиардам. В процессе онтогенеза ДНК постоянно подвергается действию различных факторов, повреждающих структуру ДНК. Относительная стабильность генетического материала связана не со свойством ее консервативности, а с наличием во всех клетках живого организма систем репарации, устраняющих возникающие повреждения ДНК. 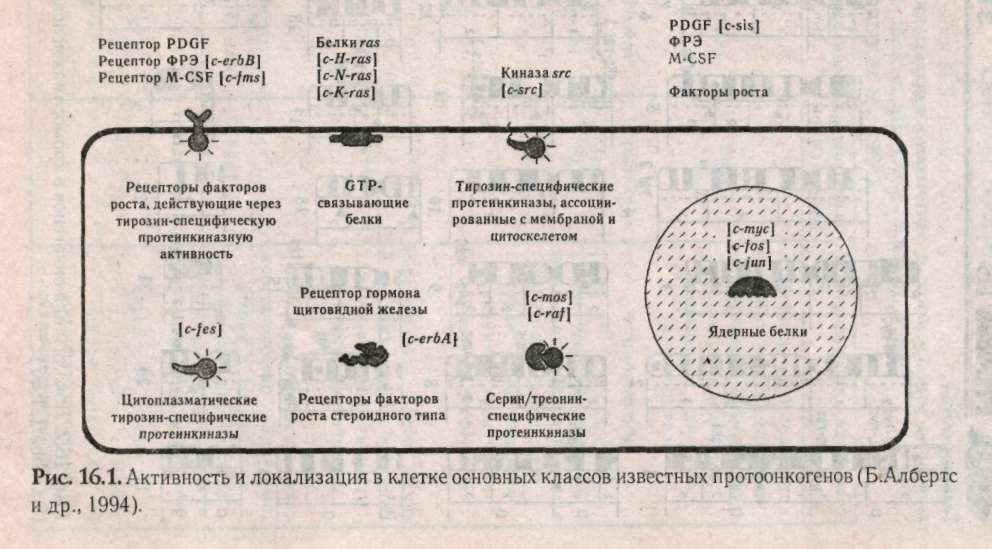 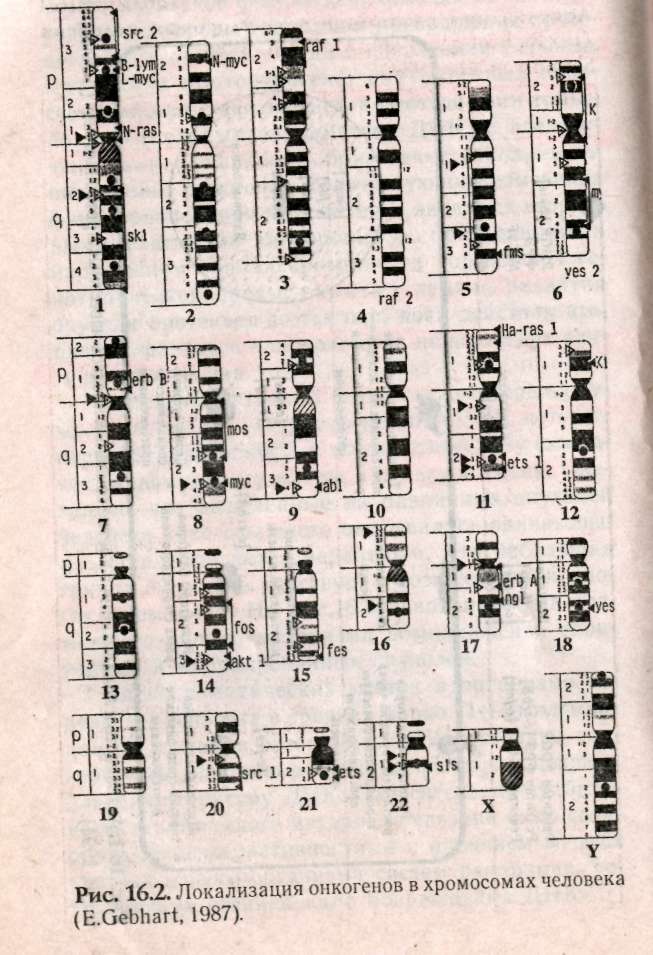 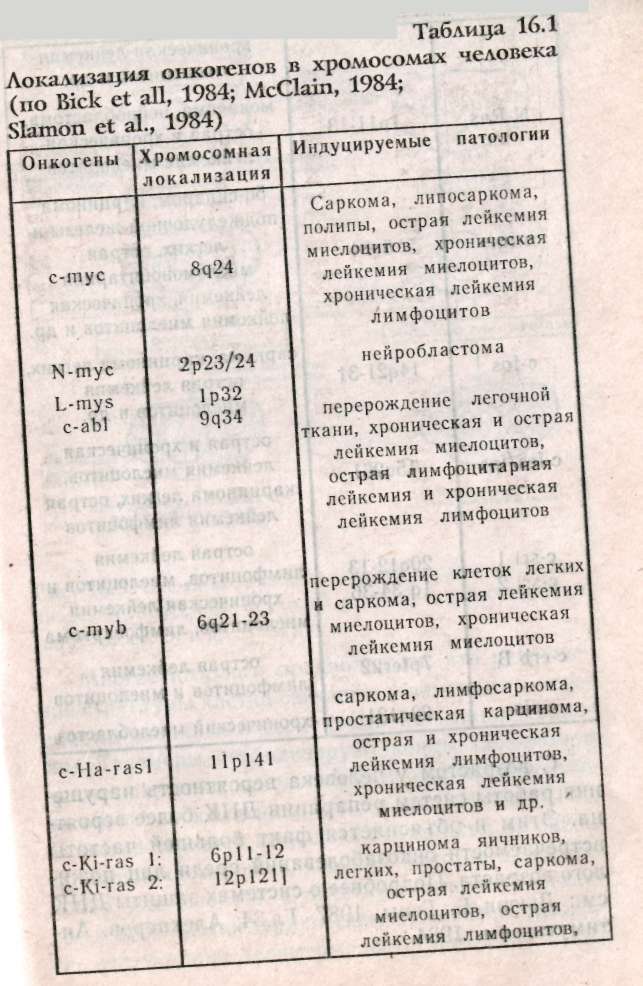  С возрастом у человека вероятность нарушения работы систем репарации ДНК более вероятна. Этим и объясняется факт большей частоты встречаемости онкозаболеваний среди лиц пожилого возраста. Подробнее о системах защиты ДНК см.: Льюин Б. Гены. 1987. Гл.34; Алекперов. Антимутагены. 1984. 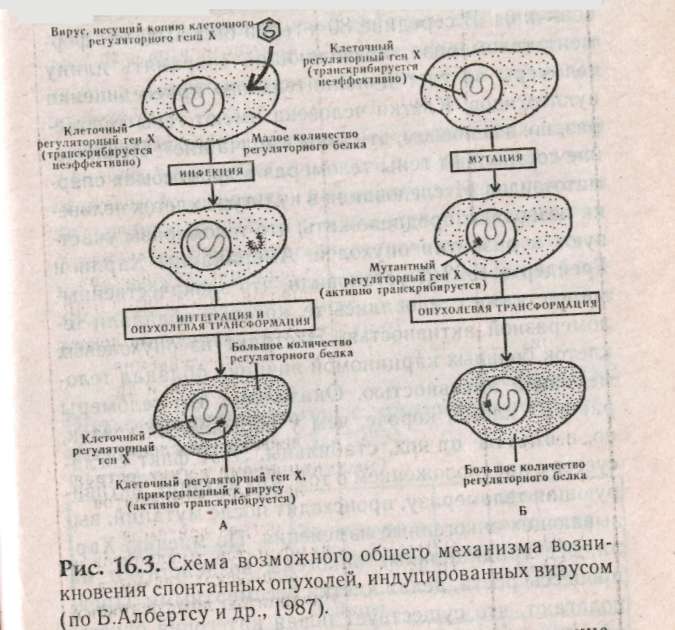 Многие цитологи предполагают, что разрушение структуры клетки связано с теломерами — специализированными сегментами на концах хромосом. Теломеры стабилизируют концы хромосом и предохраняют их от слипания и разрушения. Когда во время клеточного цикла удваивается ДНК, то всегда теряется несколько субъединиц на одном из концов хромосом. С каждым митозом длина теломер уменьшается. Существует гипотеза, согласно которой клетки теряют свою способность делиться, когда длина теломеры достигает критической 334  Высказывается мнение о мультифакторном наследовании некоторых онкологических заболеваний (карцинома молочной железы, желудка и др.), так как они часто встречаются в отдельных семьях. Однако одни только генетические факторы не могут объяснить предрасположенности к онкологическим заболеваниям, нельзя отрицать и роль среды (например, рак легких связан с курением, хотя не все курильщики заболевают). Вероятно, в конечном итоге появление опухолей обусловлено взаимодействием множества генетических механизмов и факторов среды. |