Патфиз ч.1. Патфиз ч. Гл. 1 Введение в предмет Гл. 2 Общая нозология
 Скачать 9.21 Mb. Скачать 9.21 Mb.
|

|
Углеводы — обязательный и наиболее значительный компонент пищи. В сутки человек потребляет 400–600 г различных углеводов. Как необходимый участник метаболизма, углеводы включены практически во все виды обмена веществ: нуклеиновых кислот (в виде рибозы и дезоксирибозы), белков (например, гликопротеинов), липидов (например, гликолипидов), нуклеозидов (например, аденозина), нуклеотидов (например, АТФ, АДФ, АМФ), ионов (например, обеспечивая энергией их трансмембранный перенос и внутриклеточное распределение). Как важный компонент клеток и межклеточного вещества, углеводы входят в состав структурных белков (например, гликопротеинов), гликолипидов, гликозаминогликанов и других. Как один из главных источников энергии, углеводы необходимы для обеспечения жизнедеятельности организма. Наиболее важны углеводы для нервной системы. Ткань мозга использует примерно 2/3 всей глюкозы, поступающей в кровь. ТИПОВЫЕ ФОРМЫ НАРУШЕНИЙ Расстройства метаболизма углеводов объединяют в несколько групп их типовых форм патологии: гипогликемии, гипергликемии, гликогенозы, гексоз и пентоземии, агликогенозы (рис. 8–1). Рис. 8–1. Типовые формы нарушения углеводного обмена. ГИПОГЛИКЕМИИ Гипогликемии — состояния, характеризующиеся снижением уровня глюкозы плазмы крови (ГПК) ниже нормы (менее 65 мг%, или 3,58 ммоль/л). В норме ГПК натощак колеблется в диапазоне 65–110 мг%, или 3,58–6,05 ммоль/л. ПРИЧИНЫ ГИПОГЛИКЕМИИ Причины гипогликемии представлены на рис. 8–2. 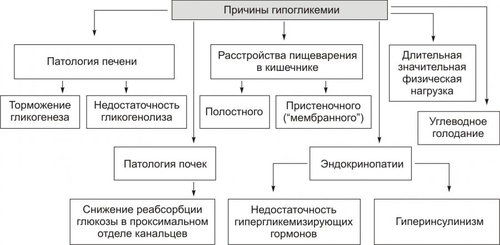 Рис. 8–2. Причины гипогликемии. ПАТОЛОГИЯ ПЕЧЕНИ Наследственные и приобретённые формы патология печени — одна из наиболее частых причин гипогликемии. Гипогликемия характерна для хронических гепатитов, циррозов печени, гепатодистрофий (в том числе иммуноагрессивного генеза), для острых токсических поражений печени, для ряда ферментопатий (например, гексокиназ, гликогенсинтетаз, глюкозо–6фосфатазы) и мембранопатий гепатоцитов. К гипогликемии приводят нарушения транспорта глюкозы из крови в гепатоциты, снижение активности гликогенеза в них и отсутствие (или малое содержание) депонированного гликогена. НАРУШЕНИЯ ПИЩЕВАРЕНИЯ Нарушения пищеварения — полостного переваривания углеводов, а также их пристеночного расщепления и абсорбции — приводят к развитию гипогликемии. Гипогликемия развивается также при хронических энтеритах, алкогольном панкреатите, опухолях поджелудочной железы, синдромах нарушенного всасывания. • Причины нарушений полостного переваривания углеводов † Недостаточность † Недостаточное содержание и/или активность амилолитических ферментов кишечника (например, при хронических энтеритах, резекции кишечника). • Причины нарушений пристеночного расщепления и абсорбции углеводов † Недостаточность дисахаридаз, расщепляющих углеводы до моносахаридов — глюкозы, галактозы, фруктозы. † Недостаточность ферментов трансмембранного переноса глюкозы и других моносахаридов (фосфорилаз), а также белка–переносчика глюкозы GLUT5. ПАТОЛОГИЯ ПОЧЕК Гипогликемия развивается при нарушении реабсорбции глюкозы в проксимальных канальцах нефрона почек. Причины: • Дефицит и/или низкая активность ферментов (ферментопатия, энзимопатия), участвующих в реабсорбции глюкозы. • Нарушение структуры и/или физикохимического состояния мембран (мембранопатии) вследствие дефицита или дефектов мембранных гликопротеинов, участвующих в реабсорбции глюкозы (подробнее см. в приложении «Справочник терминов», статья «Переносчики глюкозы» на компакт-диске). Названные причины приводят к развитию синдрома, характеризующегося гипогликемией и глюкозурией («почечный диабет»). ЭНДОКРИНОПАТИИ Основные причины развития гипогликемии при эндокринопатиях: недостаток эффектов гипергликемизирующих факторов или избыток эффектов инсулина. • К гипергликемизирующим факторам относят глюкокортикоиды, йодсодержащие гормоны щитовидной железы, СТГ, катехоловые амины и глюкагон. † Глюкокортикоидная недостаточность (например, при гипокортицизме вследствие гипотрофии и гипоплазии коры надпочечников). Гипогликемия развивается в результате торможения глюконеогенеза и дефицита гликогена. † Дефицит тироксина (T4) и трийодтиронина (T3) (например, при микседеме). Гипогликемия при гипотиреозах является результатом торможения процесса гликогенолиза в гепатоцитах. † Недостаток СТГ (например, при гипотрофии аденогипофиза, разрушении его опухолью, кровоизлиянии в гипофиз). Гипогликемия при этом развивается в связи с торможением гликогенолиза и трансмембранного переноса глюкозы. † Дефицит катехоламинов (например, при туберкулёзе с развитием надпочечниковой недостаточности). Гипогликемия при дефиците катехоламинов является следствием пониженной активности гликогенолиза. † Недостаток глюкагона (например, при деструкции • Избыток инсулина и/или его эффектов Причины гипогликемии при гиперинсулинизме: † активация утилизации глюкозы клетками организма, † торможение глюконеогенеза, † подавление гликогенолиза. Указанные эффекты наблюдаются при инсулиномах или передозировке инсулина. УГЛЕВОДНОЕ ГОЛОДАНИЕ Углеводное голодание наблюдается в результате длительного общего голодания, в том числе — углеводного. Дефицит в пище только углеводов не приводит к гипогликемии в связи с активацией глюконеогенеза (образование углеводов из неуглеводных веществ). ДЛИТЕЛЬНАЯ ЗНАЧИТЕЛЬНАЯ ГИПЕРФУНКЦИЯ ОРГАНИЗМА ПРИ ФИЗИЧЕСКОЙ РАБОТЕ Гипогликемия развивается при длительной и значительной физической работе в результате истощения запасов гликогена, депонированного в печени и скелетных мышцах. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ ГИПОГЛИКЕМИИ Возможные последствия гипогликемии (рис. 8–3): гипогликемическая реакция, синдром и кома. 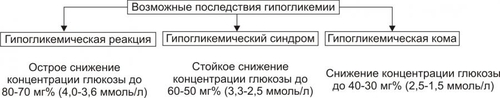 Рис. 8–3. Возможные последствия гипогликемии. ГИПОГЛИКЕМИЧЕСКАЯ РЕАКЦИЯ Гипогликемическая реакция — острое временное снижение ГПК до нижней границы нормы (как правило, до 80–70 мг%, или 4,0–3,6 ммоль/л). • Причины † Острая избыточная, но преходящая секреция инсулина через 2–3 сут после начала голодания. † Острая чрезмерная, но обратимая секреция через несколько часов после нагрузки глюкозой (с диагностической или лечебной целью, переедания сладкого, особенно у лиц пожилого и старческого возраста). • Проявления † Низкий уровень ГПК. † Лёгкое чувство голода. † Мышечная дрожь. † Тахикардия. Указанные симптомы в покое выражены слабо и выявляются при дополнительной физической нагрузке или стрессе. ГИПОГЛИКЕМИЧЕСКИЙ СИНДРОМ Гипогликемический синдром — стойкое снижение ГПК ниже нормы (до 60–50 мг%, или 3,3–2,5 ммоль/л), сочетающееся с расстройством жизнедеятельности организма. Проявления гипогликемического синдрома приведены на рис. 8–4. По происхождению они могут быть как адренергическими (обусловленными избыточной секрецией катехоламинов), так и нейрогенными (вследствие расстройств функций ЦНС). 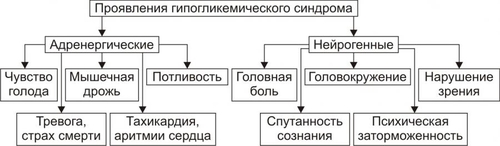 Рис. 8–4. Проявления гипогликемического синдрома. ГИПОГЛИКЕМИЧЕСКАЯ КОМА Гипогликемическая кома — состояние, характеризующееся падением ГПК ниже нормы (как правило, менее 40–30 мг%, или 2,0–1,5 ммоль/л), потерей сознания, значительными расстройствами жизнедеятельности организма. Механизмы развития • Нарушение энергетического обеспечения нейронов, а также клеток других органов вследствие: † Недостатка глюкозы. † Дефицита короткоцепочечных метаболитов свободных жирных кислот — ацетоуксусной и † Нарушения транспорта АТФ и расстройств использования энергии АТФ эффекторными структурами. • Повреждение мембран и ферментов нейронов и других клеток организма. • Дисбаланс ионов и воды в клетках: потеря ими K+, накопление H+, Na+, Ca2+, воды. • Нарушения электрогенеза в связи с указанными выше расстройствами. ПРИНЦИПЫ ТЕРАПИИ ГИПОГЛИКЕМИЙ Принципы устранения гипогликемического синдрома и комы: этиотропный, патогенетический и симптоматический ЭТИОТРОПНЫЙ Этиотропный принцип направлен на ликвидацию гипогликемии и лечение основного заболевания. Ликвидация гипогликемии Введение в организм глюкозы: • в/в (для устранения острой гипогликемии одномоментно 25–50 г в виде 50% раствора. В последующем инфузия глюкозы в меньшей концентрации продолжается до восстановления сознания у пациента). • с пищей и напитками. Это необходимо в связи с тем, что при в/в введении глюкозы не восстанавливается депо гликогена в печени (!). Терапия основного заболевания, вызвавшего гипогликемию (болезней печени, почек, ЖКТ, желёз внутренней секреции и др.). ПАТОГЕНЕТИЧЕСКИЙ Патогенетический принцип терапии ориентирован на:. • блокирование главных патогенетических звеньев гипогликемической комы или гипогликемического синдрома (расстройств энергообеспечения, повреждения мембран и ферментов, нарушений электрогенеза, дисбаланса ионов, КЩР, жидкости и других). • ликвидацию расстройств функций органов и тканей, вызванных гипогликемией и её последствиями. Устранение острой гипогликемии, как правило, приводит к быстрому «выключению» её патогенетических звеньев. Однако хронические гипогликемии требуют целенаправленной индивидуализированной патогенетической терапии. СИМПТОМАТИЧЕСКИЙ Симптоматический принцип лечения направлен на устранение симптомов, усугубляющих состояние пациента (например, сильной головной боли, страха смерти, резких колебаний АД, тахикардии и др.). ГЛИКОГЕНОЗЫ Гликогенозы — типовая форма патологии углеводного обмена наследственного или врождённого генеза, характеризующаяся накоплением избытка гликогена в клетках, что приводит к нарушению жизнедеятельности организма. Гликогенозы развиваются вследствие мутаций генов, кодирующих синтез ферментов расщепления (реже - образования) гликогена. Это приводит к отсутствию или низкой активности ферментов гликогенолиза, реже — синтеза гликогена (например, гликогеноз типа IV). Почти все гликогенозы наследуются по аутосомно-рецессивному типу. Упрощенная классификация гликогенозов (по Кори) приведена на рис. 8–5. Современное состояние вопроса (включая классификацию и проявления) см. в статье «Гликогенозы» в приложении «Справочник терминов» на компакт-диске). 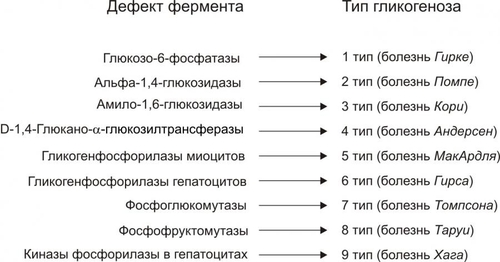 Рис. 8–5. Дефекты ферментов и основные типы гликогенозов. ГЕКСОЗЕМИИ Гексоземии — состояния, характеризующиеся увеличением содержания в крови гексоз выше нормы (более 6,4 ммоль/л, или 1,15 г/л). Наибольшую клиническую значимость имеет галактоземия и фруктоземия. ГАЛАКТОЗЕМИЯ Наиболее часто галактоземия, или галактозный диабет наследственного или врождённого генеза наблюдается у детей через несколько дней или недель после рождения (подробнее см. в статье «Галактоземия» в приложении «Справочник терминов» на компакт-диске). ФРУКТОЗЕМИЯ Фруктоземия (в том числе врождённая непереносимость фруктозы вследствие недостаточности альдолазы В) приводит к накоплению в клетках фруктозо–1–фосфата, фруктозурии, недостаточности функций печени и почек (подробнее см. в статье «Фруктоземия» в приложении «Справочник терминов» » на компакт-диске). ГИПЕРГЛИКЕМИИ Гипергликемии — состояния, характеризующиеся увеличением ГПК выше нормы (более 120 мг%, или 6,05 ммоль/л натощак). ПРИЧИНЫ ГИПЕРГЛИКЕМИИ Основными причинами гипергликемии являются эндокринопатии, неврологические и психогенные расстройства, переедание, патология печени. ЭНДОКРИНОПАТИИ Эндокринопатии — наиболее частая причина гипергликемии. Они являются следствием избытка эффектов гипергликемизирующих факторов и/или дефицита эффектов инсулина. К гипергликемизирующим гормонам относятся глюкокортикоиды, глюкагон, йодсодержащие гормоны щитовидной железы, СТГ, катехоловые амины. Избыток эффектов гипергликемизирующих факторов • Глюкагон (например, в результате гиперплазии • Глюкокортикоиды (например, при гипертрофии или опухолях коры надпочечников — кортикостеромах, болезни ИценкоКушинга) активируют глюконеогенез и ингибируют активность гексокиназы. • Катехоламины (например, при феохромоцитоме — гормонально активной опухоли мозгового вещества надпочечников) приводят к гипергликемии за счёт активации гликогенолиза. • Тиреоидные гормоны (например, при диффузном или узловом гормонально активном зобе) вызывают гипергликемию за счёт: † усиления гликогенолиза, † торможения гликогенеза из глюкозы и МК, † стимуляции глюконеогенеза, † активации всасывания глюкозы в кишечнике. • СТГ (например, при гормонально активной аденоме или опухолях аденогипофиза). Гипергликемия в условиях избытка СТГ является, в основном, результатом активации гликогенолиза и торможения утилизации глюкозы в ряде тканей. Гипергликемия может развиваться также вследствие гиперсенситизации и/или увеличения числа рецепторов к указанным выше гормрнам у клеток-мишеней. Недостаток инсулина и/или его эффектов (гипоинсулинизм). Наиболее часто гипергликемия наблюдается при СД. Гипергликемия при гипоинсулинизме является результатом: • снижения утилизации глюкозы клетками, • активации глюконеогенеза, • усиления гликогенолиза. НЕВРОЛОГИЧЕСКИЕ И ПСИХОГЕННЫЕ РАССТРОЙСТВА Нейро и психогенные расстройства (например, состояния психического возбуждения, стресс-реакции, каузалгии) характеризуются активацией симпатикоадреналовой, гипоталамо-гипофизарно-надпочечниковой и тиреоидной систем. Гормоны этих систем (катехоламины, глюкокортикоиды, T4 и T3) вызывают ряд эффектов (активация гликогенолиза, торможение гликогенеза, стимуляция глюконеогенеза), приводящих к значительной гипергликемии (механизмы смю выше). ПЕРЕЕДАНИЕ Переедание (в том числе — длительное избыточное потребление сладостей и легкоусваивающихся углеводов с пищей) — одна из причин гипергликемии. Глюкоза быстро всасывается в кишечнике. Уровень ГПК повышается и превышает возможность гепатоцитов включать её в процесс гликогенеза. Кроме того, избыток углеводсодержащей пищи в кишечнике стимулирует гликогенолиз в гепатоцитах, потенцируя гипергликемию. ПАТОЛОГИЯ ПЕЧЕНИ При печёночной недостаточности может развиваться преходящая гипергликемия в связи с тем, что гепатоциты не способны трансформировать глюкозу в гликоген. Обычно это наблюдается после приёма пищи. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ ГИПЕРГЛИКЕМИИ Возможные последствия гипергликемии: гипергликемический синдром и гипергликемическая кома (рис. 8–6). 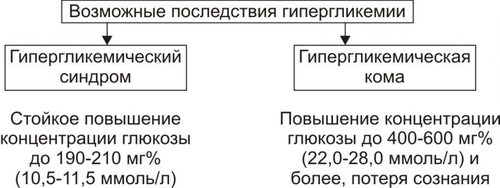 Рис. 8–6. Возможные последствия гипергликемии. ГИПЕРГЛИКЕМИЧЕСКИЙ СИНДРОМ Гипергликемический синдром — состояние характеризующееся значительным и относительно длительным увеличением ГПК выше нормы (до 190–210 мг%, т.е. 10,5–11,5 ммоль/л и более), сочетающееся с расстройством жизнедеятельности организма. Проявления • Глюкозурия. Является результатом гипергликемии. • Полиурия — повышенное мочеобразование и мочевыделение. Развивается вследствие: † повышения осмоляльности мочи, † увеличения в связи с этим клубочковой фильтрации, † снижения канальцевой реабсорбции воды. • Полидипсия — повышенное потребление жидкости. Вызывается усиленной жаждой — возникает вследствие значительной потери организмом жидкости. • Гипогидратация организма — уменьшение содержания жидкости в организме. Возникает в результпте полиурии. • Артериальная гипотензия. Она обусловлена: † гиповолемией — уменьшением объёма циркулирующей крови (ОЦК) вследствие гипогидратации организма. † уменьшением сердечного выброса крови в связи с гиповолемией. ГИПЕРГЛИКЕМИЧЕСКАЯ КОМА Гипергликемическая (гиперосмолярная) кома рассмотрена в разделе «Сахарный диабет» главы 8. ПРИНЦИПЫ УСТРАНЕНИЯ ГИПЕРГЛИКЕМИИ Основным эффективным принципом устранения гипергликемии является этиотропный. Он направлен на ликвидацию причины гипергликемии. Достижение этого и, как следствие — нормализация ГПК, обычно приводят к устранению других проявлений гипергликемии. САХАРНЫЙ ДИАБЕТ Сахарный диабет (СД) — одно из наиболее тяжелых заболеваний, чреватых тяжёлыми осложнениями, инвалидизацией и смертью. Зарегистрированная заболеваемость колеблется в разных странах от 1 до 3% (в России около 2%), а у лиц с ожирением разной степени достигает 15-25%. Ожирение, СД, артериальная гипертензия и ИБС составляют так называемый «метаболический синдром» или «смертельный квартет». По данным экспертов ВОЗ, диабет увеличивает общую смертность пациентов в 2–3 (!) раза. Примерно в 3 раза чаще у них выявляется сердечно-сосудистая патология и случаи инсульта, в 10 раз — слепота, в 20 раз — гангрена конечностей. СД — одна из причин поражений почек, ведущих к смерти пациентов. Диабет уменьшает продолжительность жизни в среднем на 7% от её общего среднего показателя.
ВИДЫ САХАРНОГО ДИАБЕТА Комитет экспертов ВОЗ по СД разработал классификацию, которая постоянно дополняется и уточняется. В ней выделяют первичные и вторичные формы СД ПЕРВИЧНЫЕ ФОРМЫ САХАРНОГО ДИАБЕТА Первичные формы СД характеризуются отсутствием у пациента какихлибо определённых заболеваний, вторично приводящих к развитию диабета. Различают две разновидности первичного СД (табл. 8–1): • инсулинзависимый сахарный диабет (ИЗСД). • инсулиннезависимый сахарный диабет (ИНСД). Таблица 8–1. Отличия инсулинзависимого (ИЗСД) и инсулиннезависимого (ИНСД) сахарного диабета
* Определения «абсолютный» и «относительный» применительно к термину «дефицит инсулина» необходимо понимать не в контексте «содержание инсулина» (например, в таких-то единицах), а в контексте термина «недостаточность эффектов инсулина» (с акцентом на слово «эффект») для поддержания параметров углеводного и других видов метаболизма. Понятие «ИЗСД» подразумевает: • Абсолютный дефицит инсулина. • Необходимость постоянного применения инсулина. • Реальную угрозу развития кетоацидоза. Пациентам с ИЗСД назначают такую дозу инсулина, которая необходима для поддержания оптимального уровня ГПК. Отмена или дефицит инсулина вызывает у них кетоацидоз. Понятие «ИНСД» подразумевает формы диабета, обусловленные недостаточностью эффектов инсулина при нормальном или даже повышенном уровне гормона в крови. • Функция • Большинство пациентов не нуждается в обязательном введении инсулина. • Расстройства жизнедеятельности организма развиваются относительно медленно. • ИНСД составляет не менее 80% всех случаев СД. ВТОРИЧНЫЕ ФОРМЫ САХАРНОГО ДИАБЕТА Вторичные формы СД являются следствием либо какойлибо основной болезни или патологического состояния, вторично повреждающих поджелудочную железу, либо воздействия на неё физических или химических факторов. К таким болезням, патологическим состояниям и воздействиям относятся: • Заболевания, поражающие ткань поджелудочной железы (например, панкреатит). • Другие болезни эндокринной системы (например, семейный полиэндокринный аденоматоз). • Действие на поджелудочную железу химических или физических агентов. САХАРНЫЙ ДИАБЕТ ТИПОВ I И II В более ранних классификациях выделяли СД типов I и II. Эти обозначения вначале применяли как синонимы ИЗСД и ИНСД соответственно. Современные специалисты считают такой подход не совсем корректным. Это объясняется тем, что, например, больные с ИНСД также могут приобрести зависимость от инсулина. При его недостатке у них развивается кетоацидоз, чреватый коматозным состоянием (например, это наблюдается у многих пациентов без ожирения, имеющих в крови АТ к • Термин «Тип I СД» применяли для обозначения тех его вариантов, основным патогенетическим звеном которых являлся иммунный (иммуноагрессивный) механизм. Диабет типа I наблюдают у 10–15% пациентов, страдавших СД. • Термин «Тип II СД» рекомендовали использовать для той формы СД, патогенез которой не включал в качестве причинного (!) иммунный механизм. СД типа II диагностировали более чем у 85% пациентов с СД. Таким образом, СД развивается в результате либо: • дефицита инсулина (т.е. в результате гипоинсулинизма или абсолютной инсулиновой недостаточности), либо: • недостаточности эффектов инсулина при его нормальном или даже повышенном содержании в плазме крови. ЭТИОЛОГИЯ САХАРНОГО ДИАБЕТА СД развивается либо вследствие дефицита инсулина (ИЗСД), либо недостаточности его эффектов (ИНСД). ПРИЧИНЫ • Дефицит инсулина может возникнуть под влиянием факторов биологической, химической, физической природы, а также при воспалительных процессах поджелудочной железы † Биологические факторы ‡ Генетические дефекты ‡ Иммунные факторы. Иммуноглобулины, цитотоксические T-лимфоциты, а также продуцируемые ими цитокины способны повреждать § к цитоплазматическим Аг — IСА (от англ. islet cell autoantibody — аутоантитела к белкам островковых клеток); § к белку с мол. массой 64 кД, обнаруживаемому в цитоплазматической мембране § к молекулам инсулина. ‡ Вирусы, тропные к § прямое цитолитическое действие в отношение § инициирование иммунных процессов в адрес § развитие воспаления в участках расположения ‡ Эндогенные токсические вещества, повреждающие † Химические факторы: аллоксан, высокие дозы этанола, цитостатики и другие ЛС (например, противоопухолевый препарат стрептозоцин). † Физические факторы: проникающая радиация, инициирующая активацию липопероксидных процессов; механическая травма поджелудочной железы, сдавление опухолью. Указанные и другие факторы физической природы приводят к гибели островковых † Воспалительные процессы Воспалительные процессы, возникающие в поджелудочной железе под действием факторов биологической (главным образом, микроорганизмов), химической и физической природы. Хронические панкреатиты примерно в 30% случаев являются причиной инсулиновой недостаточности. • Недостаточность эффектов инсулина развивается под влиянием причин нейро- или психогенных природы, контринсулярных факторов, а также вследствие дефектов инсулиновых рецепторов и пострецепторных нарушений в клетках-мишенях (рис. 8–7) 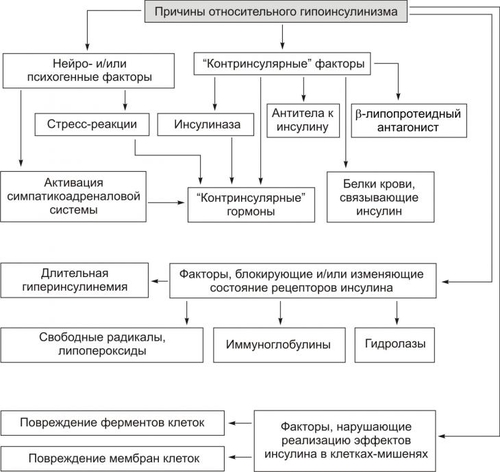 Рис. 8–7. Причины относительного гипоинсулинизма. † Нейро и/или психогенные факторы ‡ Активация нейронов ядер заднего гипоталамуса, приводящая к повышению тонуса симпатикоадреналовой и гипоталамогипофизарнонадпочечниковой системы. Это обусловливает значительное и стойкое увеличение в крови контринсулярных гипергликемизирующих гормонов: адреналина, норадреналина (надпочечникового генеза), глюкокортикоидов и, следовательно, относительную недостаточность эффектов инсулина. ‡ Повторное развитие затяжных стресс–реакций. Последние включают активацию симпатикоадреналовой и гипоталамо–гипофизарно–надпочечниковой систем. Это приводит к повышению содержания в крови катехоламинов, глюкокортикоидов, тиреоидных гормонов. † Контринсулярные факторы ‡ Чрезмерная активация инсулиназы гепатоцитов. Эта протеаза гидролизует молекулы инсулина. ‡ АТ к эндогенному инсулину. ‡ Повышение содержания в крови контринсулярных (гипергликемизирующих) гормонов: катехоламинов, глюкагона, глюкокортикоидов, СТГ, Т3 и Т4. Гиперпродукция указанных гормонов может наблюдаться при опухолях соответствующих эндокринных желёз или при длительном стрессе. ‡ Повышенная концентрация в плазме крови белков, связывающих молекулы инсулина. † Факторы, вызывающие блокаду, деструкцию или снижение чувствительности рецепторов инсулина: ‡ Ig, имитирующие структуру молекулы инсулина. Такие Ig взаимодействуют с рецепторами инсулина, блокируют их, закрывая тем самым доступ к рецептору молекулам инсулина. ‡ Ig, разрушающие рецепторы инсулина и/или перирецепторную зону клеток–мишеней. ‡ Длительный избыток инсулина, вызывающий гипосенситизацию клеток–мишеней к гормону. ‡ Гидролазы, высвобождающиеся из лизосом и активирующиеся внутри и вне повреждённых или разрушающихся клеток (например, при общей гипоксии, расстройствах внешнего дыхания и кровообращения). ‡ Свободные радикалы и продукты СПОЛ (например, при повторном затяжном стрессе, атеросклерозе, сердечно-сосудистой недостаточности). † Факторы, нарушающие реализацию эффектов инсулина в клетках-мишенях: ‡ повреждающие мембраны и/или рецепторы клеток к инсулину. ‡ денатурирующие и/или разрушающие клеточные ферменты. Наиболее частыми причинами повреждения мембран и ферментов клеток являются избыточная активность лизосомальных ферментов, чрезмерное образование активных форм кислорода, свободных радикалов и гидроперекисей липидов. Эти и другие патогенные агенты подавляют транспорт глюкозы в клетки, образование цАМФ, трансмембранный перенос ионов Ca2+ и Mg2+, необходимых для реализации внутриклеточных эффектов инсулина. ФАКТОРЫ РИСКА Известно большое число факторов риска развития СД. • Избыточная масса тела. Ожирение выявляется более чем у 80% пациентов с ИНСД. Это повышает инсулинорезистентность печени, жировой и других тканей–мишеней инсулина. • Стойкая и значительная гиперлипидемия. Оба фактора стимулируют продукцию контринсулярных гормонов и гипергликемию. Это, в свою очередь, активирует синтез инсулина • Артериальная гипертензия, приводящая к нарушению микроциркуляции в поджелудочной железе. • Наследственная или врождённая предрасположенность. Считают, что у пациентов с иммуноагрессивным диабетом предрасположенность к болезни определяют гены HLA. У пациентов с ИНСД предрасположенность к диабету имеет полигенный характер. При наличии СД у одного из родителей соотношение их больных детей к здоровым может составлять 1:1. • Женский пол. • Повторные стресс–реакции. Они сопровождаются стойким повышением уровней в крови контринсулярных гормонов. • Сочетание нескольких факторов риска увеличивает вероятность возникновения диабета в 20-30 раз. ПАТОГЕНЕЗ САХАРНОГО ДИАБЕТА Ниже рассмотрены звенья патогенеза СД при дефиците инсулина (в результате чего развивается ИЗСД) и при недостаточности эффектов инсулина (в связи с чем развивается ИНСД). ДЕФИЦИТ ИНСУЛИНА Основные звенья патогенеза абсолютной инсулиновой недостаточности приведены на рис. 8–8. 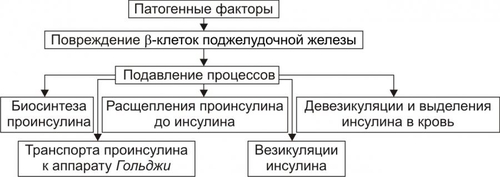 Рис. 8–8. Основные звенья патогенеза абсолютной инсулиновой недостаточности. • При дефиците инсулина происходит: † повреждение и гибель † уменьшение суммарной массы † подавление синтеза и выделения в кровь инсулина из повреждённых • В большинстве случаев (возможно, даже во всех) патогенез инсулиновой недостаточности имеет общее звено: развитие иммуноагрессивного процесса. Этот процесс обычно длится несколько лет и сопровождается постепенной деструкцией † Симптомы диабета, как правило, появляются при разрушении примерно 75–80% † У погибших от СД пациентов масса поджелудочной железы составляет в среднем 40 г (при 80–85 г в норме). При этом масса 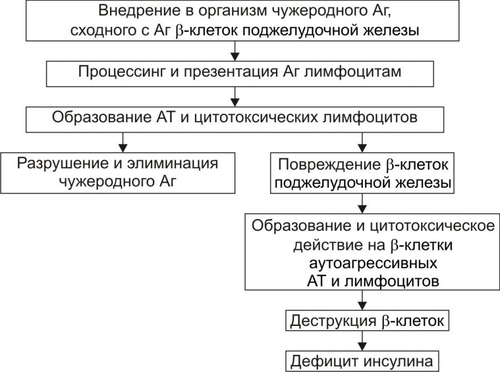 Рис. 8–9. Основные звенья иммуноагрессивного варианта патогенеза сахарного диабета. • К числу главных звеньев механизма развития иммуноагрессивного варианта СД относятся следующие (рис. 8–9): † Внедрение в организм генетически предрасположенных к СД лиц носителя чужеродного Аг. Наиболее часто это вирусы, реже — другие микроорганизмы. † Поглощение чужеродного Аг антигенпредставляющими клетками, процессинг Аг и представление его в сочетании с Аг HLA (презентация) хелперным T-лимфоцитам. † Образование специфических АТ и активированных лимфоцитов против чужеродного Аг. † Действие АТ и активированных лимфоцитов на: ‡ чужеродный Аг. Это обеспечивает его разрушение и элиминацию из организма при участии фагоцитов; ‡ антигенные структуры † Поглощение, процессинг и презентация лимфоцитам как чужеродных Аг, так и вновь образовавшихся аутоантигенов Процесс иммунной аутоагрессии потенцируется синтезом и транспортом на поверхность повреждённых † Миграция в регионы повреждённых и разрушенных † Реализация цитолитического эффекта лейкоцитов на † Высвобождение из разрушеннных † Поглощение макрофагами указанных цитоплазматических белков Признаки активации системы иммунного надзора по отношению к • Патогенез абсолютной инсулиновой недостаточности, вызванной действием химических панкреотропных факторов, рассмотрен на рис. 8–10. 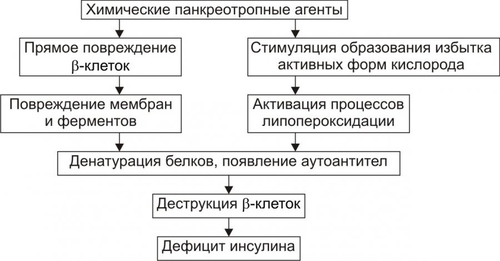 Рис. 8–10. Основные звенья патогенеза сахарного диабета при действии химических панкреотропных агентов. • Механизм развития абсолютной инсулиновой недостаточности, вызванной влиянием физических патогенных факторов, представлен на рис. 8–11. 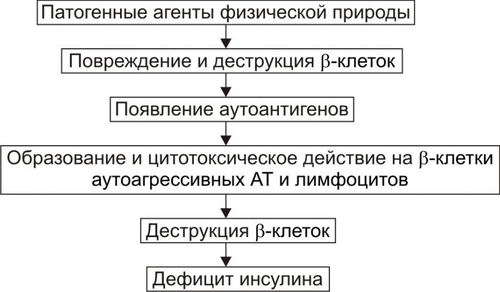 Рис. 8–11. Основные звенья патогенеза сахарного диабета при действии физических патогенных факторов. НЕДОСТАТОЧНОСТЬ ЭФФЕКТОВ ИНСУЛИНА Реализация различных вариантов патогенеза СД при недостаточности эффектов инсулина происходит при нормальном или даже повышенном его синтезе и инкреции в кровь (при этом развивается ИНСД). • Контринсулярные факторы † Инсулиназа ‡ Механизмы активации инсулиназы: § увеличение содержания в крови глюкокортикоидов и/или СТГ (что нередко наблюдается у пациентов с СД), § дефицит ионов цинка и меди, в норме снижающих активность инсулиназы. Учитывая, что инсулиназа начинает интенсивно синтезироваться гепатоцитами в пубертатном периоде, этот механизм является одним из важных звеньев патогенеза юношеского диабета. † Протеолитические ферменты. Они могут поступать из обширных очагов воспаления и разрушать инсулин (например, при флегмоне, перитоните, инфицировании ожоговой поверхности). † АТ к инсулину крови. † Вещества, связывающие молекулы инсулина и тем самым блокирующие взаимодействие инсулина с рецепторами. К ним относятся: ‡ Плазменные ингибиторы инсулина белковой природы (например, отдельные фракции Инсулин, связанный с плазменными белками, не проявляет своей активности во всех тканях, исключая жировую. В последней создаются условия для отщепления белковой молекулы и контакта инсулина со специфическими рецепторами. ‡ • Устранение или снижение эффектов инсулина на ткани–мишени Устранение или снижение эффектов инсулина на ткани–мишени достигается благодаря гипергликемизирующему эффекту избытка гормонов — метаболических антагонистов инсулина. К ним относятся катехоламины, глюкагон, глюкокортикоиды, СТГ и йодсодержащие тиреоидные гормоны. Длительная и значительная гипергликемия стимулирует повышенное образование инсулина • Инсулинорезистентность Инсулинорезистентность характеризуется нарушением реализации эффектов инсулина на уровне клеток–мишеней. Известны рецепторные и пострецепторные механизмы этого феномена. † Рецепторные механизмы ‡ «Экранирование» (закрытие) инсулиновых рецепторов Ig. Последние специфически реагируют с белками самих рецепторов и/или перирецепторной зоны. Молекулы Ig при этом делают невозможным взаимодействие инсулина и его рецептора. ‡ Гипосенситизация клеток-мишеней к инсулину. Она обусловлена длительным повышением концентрации инсулина в крови и в интерстиции. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||