Коммуникации эндокринная
 Скачать 1.15 Mb. Скачать 1.15 Mb.
|

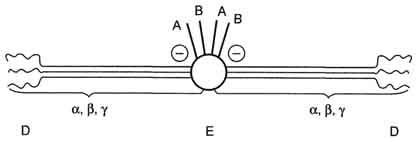 Рис. 14-8. Строение фибриногена. Фибриноген состоит из шести полипептидных цепей: Аα2, Вβ2 и γ2. А, В - отрицательнозаряженные фрагменты, благодаря которым молекулы фибриногена не агрегируют. Д, E - глобулярные домены молекулы фибриногена. Домены отделены участками полипептидных цепей, имеющими стержнеобразную конфигурацию. Из центрального глобулярного домена E выступают N- концевые участки фрагментов А и В цепей Аα2 и Вβ2. а две другие - В с β-цепью в Аα2- и Вβ2-цепях фибриногена. Мономер фибрина, образующийся из фибриногена, имеет состав (α, β, γ)2. Образование нерастворимого геля фибрина. На втором этапе образуется нерастворимый полимерный фибриновый сгусток - гель фибрина. В результате превращения фибриногена в фибрин-мономер в домене E открываются центры связывания с доменами D. Причём домен E содержит центры агрегации, формирующиеся только после частичного протеолиза фибриногена под действием тромбина, а домен D является носителем постоянных центров агрегации. Первичная агрегация молекул фибрина происходит в результате взаимодействия центров связывания домена E одной молекулы с комплементарными им участками на доменах D других молекул.Таким образом, между доменами молекул фибрина-мономера образуются нековалентные связи. При "самосборке" геля фибрина сначала образуются двунитчатые протофибриллы, в которых молекулы фибрина смещены друг относительно друга на 1/2 длины. После достижения протофибриллами определённой критической длины начинается их латеральная ассоциация, ведущая к образованию толстых фибриновых волокон (рис. 14-9). Образовавшийся гель фибрина непрочен, так как молекулы фибрина в нём связаны между собой нековалентными связями. Стабилизация геля фибрина. В результате образования амидных связей между остатками лизина одной молекулы фибрина и остатками глутамина другой молекулы гель фибрина стабилизируется. Реакцию трансамидирования катализирует фермент трансглутамидаза (фактор ХIIIа) (рис. 14-10). Фактор XIII активируется частичным протеолизом под действием тромбина. Трансглутамидаза также образует амидные связи между фибрином и фибронектином - гликопротеином межклеточного матрикса и плазмы крови (см. раздел 15). Таким образом, тромб фиксируется в месте повреждения сосуда. 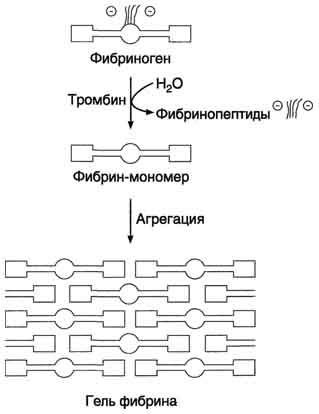 Рис. 14-9. Образование геля фибрина. Фибриноген, освобождаясь под действием тромбина от отрицательно заряженных фрагментов (фибринопептидов 2А и 2В), превращается в фибрин-мономер. В результате взаимодействия комплементарных участков E- и D-доменов фибрина-мономера происходит сначала линейная, а затем латеральная полимеризация молекул с образованием геля фибрина. 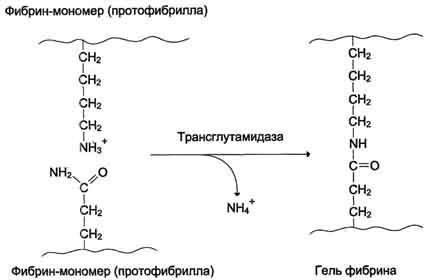 Рис. 14-10. Образование амидной связи между молекулами фибрина. Ретракция фибринового сгустка. Сжатие (ретракцию) геля обеспечивает актомиозин тромбоцитов - сократительный белок тромбостенин, обладающий АТФ-азной активностью. Тромбостенин участвует также в активации и агрегации тромбоцитов. Ретракция кровяного сгустка предупреждает полную закупорку сосудов, создавая возможность восстановления кровотока. В механизме образования тромба есть три функционально разных этапа: прокоагулянтный путь, контактный путь и антикоагулянтная фаза, препятствующая распространению тромба.  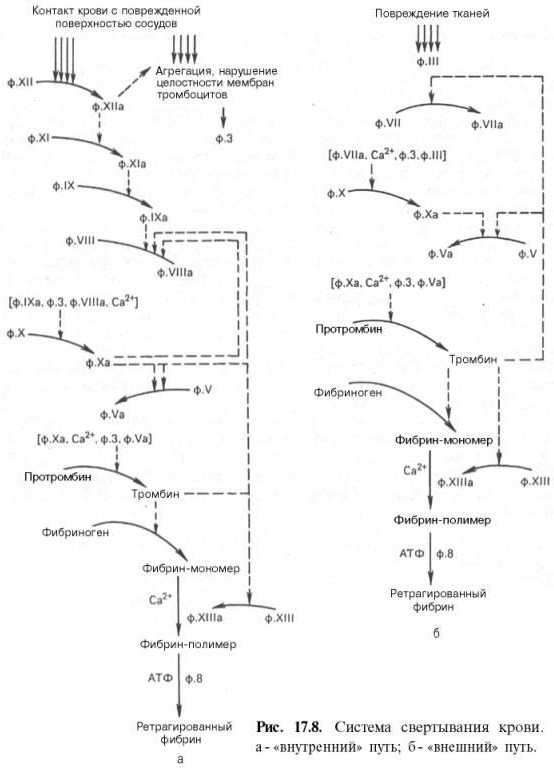 Внутренний и внешний пути свёртывания. Витамин К в свёртывании крови. Внутренний и внешний пути свёртывания. Витамин К в свёртывании крови.Витамин К и система свертывания крови Биологическая роль витамина К обусловлена участием его в процессе свертывания крови. Он синтезирует в печени активные формы протромбина - вещества, обеспечивающего нормальное свертывание крови. свёртывание крови (протромбин (фактор II), факторы VII, IX, X, белок C, белок S и белок Z). Противосвёртывающая система крови. Физиологические ингибиторы свёртывания крови играют важную роль в поддержании гемостаза, так как они сохраняют кровь в жидком состоянии и препятствуют распространению тромба за пределы повреждённого участка сосуда. Тромбин, образующийся в результате реакций прокоагулянтного и контактного путей свёртывания крови, вымывается током крови из тромба. Он может инактивироваться при взаимодействии с ингибиторами ферментов свёртывания крови или активировать антикоагулянтную фазу, тормозящую образование тромба. Антикоагулянтная фаза. Свёртывание крови должно быть ограничено не только в пространстве, но и во времени. Антикоагулянтная фаза ограничивает время существования активных факторов в крови и инициируется самим тромбином. Следовательно, тромбин, с одной стороны, ускоряет свёртывание крови, являясь последним ферментом каскада реакций коагуляции, а с другой - тормозит его, вызывая образование ферментных комплексов антикоагулянтной фазы на неповреждённом эндотелии сосудов. Этот этап представляет собой короткий каскад реакций, в котором кроме тромбина участвуют белок-активатор тромбомодулин (Тм), витамин К-зависимая сериновая протеаза протеин С, белок-активатор S и факторы Va и VIIIa (рис. 14-16). В каскаде реакций антикоагулянтной фазы последовательно образуются 2 мембранных комплекса IIа-Тм- Са2+ и Ca-S-Са2+. Тромбомодулин - интегральный белок мембран эндотелиальных клеток. Он не требует протеолитической активации и служит белком-активатором тромбина. Тромбин приобретает способность активировать протеин С только после взаимодействия с тромбомодулином, причём связанный с тромбомодулином тромбин не может превращать фибриноген в фибрин, не активирует фактор V и тромбоциты. Протеин С - профермент, содержащий остатки γ-карбоксиглутамата. Тромбин в мембранном комплексе IIа- Тм-Са2+ активирует частичным протеолизом протеин С. Активированный протеин С (Са) образует с белком- активатором S мембраносвязанный 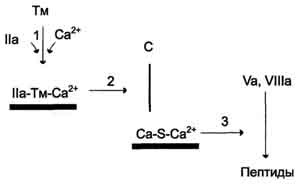 Рис. 14-16. Антикоагулянтная фаза. Тм - тромбомодулин; С - протеин С; Са - активный протеин С; S - протеин S; жирные линии - мембранно-связанный комплекс. 1 - тромбин (На) образует мембранный комплекс с белком тромбомодулином (Тм); 2 - тромбин в составе мембранного комплекса Иа-Тм-Са2+ активирует протеин С; 3 - активированный протеин С в составе ферментного мембранного комплекса Са-S- Са2+ гидролизует по 2 пептидные связи в факторах Va и VIIIa и превращает их в неактивные пептиды. комплекс Ca-S-Са2+. Са в составе этого комплекса гидролизует в факторах Va и VIIIa по две пептидные связи и инактивирует эти факторы. Под действием комплекса Ca-S-Са2+ в течение 3 мин. теряется 80% активности факторов VIIIa и Va. Таким образом, тромбин по принципу положительной обратной связи не только ускоряет своё образование, но и, активируя протеин С, тормозит процесс свёртывания крови. Наследственный дефицит протеина С и S ведёт к снижению скорости инактивации факторов VIIIa и Va и сопровождается тромботической болезнью. Мутация гена фактора V, при которой синтезируется фактор V, резистентный к протеину С, также приводит к тромбогенезу. Антикоагулянтная фаза вызывает торможение каскада реакций свёртывания крови, а ингибиторы ферментов свёртывания инактивируют активные ферменты в кровяном русле. Ингибиторы ферментов свёртывания крови. Физиологические ингибиторы ферментов свёртывания крови ограничивают распространение тромба местом повреждения сосуда. Белок плазмы крови антитромбин III - наиболее сильный ингибитор свёртывания крови; на его долю приходится около 80-90% антикоагулянтной активности крови. Он инактивирует ряд сериновых протеаз крови: тромбин, факторы IХа, Ха, ХIIа, калликреин, плазмин и урокиназу. Антитромбин III не ингибирует фактор VIIIa и не влияет на факторы в составе мембранных комплексов, а устраняет ферменты, находящиеся в плазме крови, препятствуя распространению тромбо-образования в кровотоке. Взаимодействие антитромбина с ферментами свёртывания крови ускоряется в присутствии гепарина. Гепарин - гетерополисахарид, который синтезируется в тучных клетках. В результате взаимодействия с гепарином антитромбин III приобретает конформацию, при которой повышается его сродство к сериновым протеазам крови. После образования комплекса антитромбин III-гепарин-фермент гепарин освобождается из него и может присоединяться к другим молекулам антитромбина. При наследственном дефиците антитромбина III в молодом возрасте наблюдают тромбозы и эмболии сосудов, опасные для жизни. α2-Макроглобулин образует комплекс с сериновыми протеазами крови. В таком комплексе их активный центр полностью не блокируется, и они могут взаимодействовать с субстратами небольшого размера. Однако высокомолекулярные субстраты, например фибриноген, становятся недоступными для действия протеаз в комплексе α2-макроглобулинтромбин. Антиконвер гин (тканевый ингибитор внешнего пути свёртывания) синтезируется в эндотелии сосудов. Он специфически соединяется с ферментным комплексом Тф-VIIа-Са2+, после чего улавливается печенью и разрушается в ней. α1-Антитрипсин ингибирует тромбин, фактор ХIа, калликреин, однако он не рассматривается как важный ингибитор факторов свёртывания крови, α1-Антитрипсин в основном на тканевом уровне ингибирует панкреатические и лейкоцитарные протеазы, коллагеназу, ренин, урокиназу. Пептиды, образующиеся в результате протеолитической активации проферментов и профакторов, тоже обладают выраженными антикоагулянтными свойствами, но механизм их действия в настоящее время не выяснен. Нарушения коагуляционного гемостаза:гемофилии. Наследственных коагулопатии доминируют (около 97%) гемофилии А и В В основе обоих видов гемофилии лежит мутация локусов синтеза фактора VIII (гемофилия А) или фактора IX (гемофилия В) в Х-хромосоме. Болезнь при обеих формах наследуется по рецессивному, сцепленному с полом типу; носителями болезни являются женщины, а больными — только лица мужского пола (исключения из этого правила крайне редки). Все дочери больного гемофилией, получившие патологическую Х-хромосому от отца, являются кондукторами болезни, а сыновья женщин-носительниц гемофилии в 50% случаев имеют шанс стать больными, дочери (в 50% случаев) — носителями патологического гена. Для гемофилии характерны кровоточивость гематомного типа — болезненные кровоизлияния в крупные суставы (гемартрозы), мышцы, забрюшинную клетчатку, в область черепа, гематурия, тяжелые отсроченные посттравматические и послеоперационные кровотечения, в том числе при малых травмах и вмешательствах (при порезах, удалении зубов и т. п.). Поскольку факторы VIII и IX участвуют только во внутреннем механизме свертывания крови, при гемофилии удлинены общее время свертывания цельной крови, время свертывания рекальцифицированной цитратной плазмы и активированного парциального тромбопластинового времени (АПТВ), тогда как протромбиновый показатель и тромбиновое время свертывания остаются нормальными. К группе наследственных коагулопатий относят также болезнь Виллебранда, при которой (см. выше) тромбоцитопатия сочетается с дефицитом фактора VIII. В этом случае заболевание проявляется петехиально- гематомной кровоточивостью в связи с сочетанным нарушением адгезивно-агрегационных свойств тромбоцитов и снижением коагуляционной активности крови. Это обусловлено тем, что фактор Виллебранда является переносчиком плазменного фактора VIII. При отсутствии фактора Виллебранда фактор VIII подвергается ускоренному разрушению в крови, что и обусловливает его дефицит и связанную с ним спонтанную гематомную кровоточивость. |