2-курс ПЗ рус блог. Менделевское наследование у человека
 Скачать 437.9 Kb. Скачать 437.9 Kb.
|

                     Задание 2. Существует постоянно обновляющаяся электронная база данных "Менделевское наследование у человека" (OMIM-Online Mendelian Inheritance in Man). OMIM - это всеобъемлющий, авторитетный сборник человеческих генов и генетических фенотипов, который свободно доступен и обновляется ежедневно. Полнотекстовые обзоры в OMIM содержат информацию обо всех известных болезнях с менделирующим типом наследования и более чем 15000 генов. OMIM фокусируется на взаимосвязи между фенотипом и генотипом. Эта база данных была начата в начале 1960-х годов в Университете Джона Хопкинса группой учёных и редакторов под руководством Виктора Маккьюсика в качестве каталога менделевских признаков и расстройств, озаглавленной Менделевское наследование человека (MIM). Онлайн-версия OMIM была создана в 1985 году и стала широко доступна в Интернете с 1987 года. База находится в Национальном центре биотехнологической информации (США). Ее адрес в Интернете: https://www.omim.org/. Для каждой болезни здесь суммированы клинические и молекулярно-генетические данные (о картировании, идентификации гена, практических возможностях генодиагностики). В базe данных OMIM приводится код, указывающий на тип наследования, тип мутации и связанные с ней хромосомы. Например, диапазон кода заболевания в зависимости от типа наследования следующий:
По названию болезни или комплексу симптомов можно узнать в какой хромосоме, в каком локусе находится мутантный ген, его обозначение и полное описание, ссылки на проводимые исследования по этому гену и многое другое. Практическое занятие № 2 6.1. Тема: Современные методы диагностики наследственных болезней Информационно-дидактический блок В настоящее время для диагностики наследственных болезней используются клинико-генеалогический, цитогенетические, молекулярно-генетические и биохимические методы. Клинико-генеалогический метод – один из старых методов изучения генетики человека. Предложен Ф. Гальтоном в конце 19 века. Позволяет установить наследственный характер и тип наследования признака, его пенетрантность, рассчитать риск рождения больного ребенка в семье. Суть этого метода заключается в выявлении родословных связей и прослеживании признака (болезни) среди родственников в ряду поколений. Состоит из этапов:
Сбор сведений начинается с обратившегося за консультацией лица (пробанда). На этом этапе задача сводится к наиболее полному сбору информации о каждом члене семьи путем опроса, личного осмотра и, при необходимости, специальных лабораторных исследований. Родословная составляется в виде графической схемы с помощью специальных символов (Рис.1). Поколения в родословной обозначаются римскими цифрами сверху вниз, члены одного поколения - слева направо арабскими цифрами. Братья и сестры (сибсы) располагаются в порядке рождения. Т.о., каждый член родословной имеет свой шифр, например: I-2, II-5. 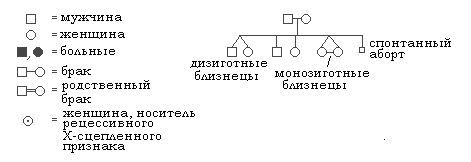 Рис. 1 Символы родословной Генеалогический анализ проводится с целью: 1 - установление наследственного характера признака 2 - определение типа наследования признака. Различают несколько основных типов наследования, которые соответственно проявляются в родословных. Аутосомно-доминантный тип (АД):
АД болезни характеризуются большим разнообразием экспрессивности признаков и сроков проявления болезни даже в пределах одной семьи. В большинстве случаев - это патологические состояния, не наносящие серьезного ущерба здоровью и репродуктивной функции - синдром Марфана, полидактилия, брахидактилия и т.д. У гомозигот, часто, болезнь проявляется в более тяжелой форме (иногда летально). Аутосомно-рецессивный тип (АР):
АР болезни составляют большую часть генетического груза популяции людей. Проявляются только у гомозигот (аа). Гетерозиготы (Аа) клинически здоровы, но являются носителями патологического гена и могут передавать его детям. Малодетность современных семей затрудняет установление рецессивных болезней. Большинство АР болезней значительно снижают жизнеспособность организма и характеризуются высокой смертностью (фенилкетонурия, галактоземия, болезнь Вильсона-Коновалова и другие формы ферментопатий, олигофрения и т.д.). Х-сцепленный-доминантный тип (ХД):
ХД болезни очень вариабельны по экспрессивности у женщин (витамино-устойчивый рахит, гипоплазия эмали). Х-сцепленный-рецессивный тип (ХР):
При данном типе наследования женщины чаще гетерозиготны и фенотипически нормальны (гемофилия, дальтонизм) У-сцепленный тип (У) - голандрическое наследование – признак проявляется только у мужчин (синдактилия, гипертрихоз). Цитоплазматическое наследование характерно для генов, локализованных в ДНК пластид и митохондрий. Описаны различные мутации генов митохондрий, вызывающие наследственные болезни у человека. Известно около 100 таких болезней: атрофия зрительного нерва Лебера, онкоцитома (доброкачественныя опухоль), митохондриальная миопатия, отсроченная кардиопатия (тяжелое заболевание молодого и среднего возраста). Основные симптомы митохондриальных болезней связаны с поражением нервной системы, характерно раннее начало. Основной диагностический тест - феномен гетероплазии митохондрий - слипание митохондрий и неравномерное их распределение в цитоплазме. Наследование митохондриальных болезней характеризуется следующими особенностями: - болезнь передается только от матери; - болеют оба пола; - больные отцы не передают болезнь детям. Полигенное наследование- критерии полигенного наследования впервые обобщены К.Картером в 1969 г. и дополнены Ф.Фогелем и А.Мотульски в 1989 г. Являются болезнями наследственной предрасположенности, зависящими от условий среды. Конкордантность монозиготных близнецов по полигенным болезням в 4 раза выше конкордантности дизиготных близнецов. Для полигенных болезней характерно: 1. Сегрегация в семьях, хотя при этом нет четкой модели наследования. 2. Зависимость риска заболевания от: - степени родства с больным пробандом - чем выше степень родства, тем выше риск заболевания; - числа больных родственников: - если оба родителя здоровы - риск 5-10 %; - если один родитель болен - риск 10-20 %; - оба родителя больны - риск до 40 %; - от редкопоражаемого пола - чем чаще болезнь проявляется у одного пола, тем выше риск для редкопоражаемого пола; - от наследуемости заболевания - чем большее число генов влияет на болезнь, тем выше риск. 3. Расчет риска рождения больного ребенка. Для этого необходимо знать: 1 - гомо- или гетерозиготен пробанд; 2 - пенетрантность признака. Пенетрантностьзависит от генетического окружения и факторов внешней среды. Ее можно рассчитать по родословной и с помощью формулы (по пробандной конкордантности). Пример расчета: В браке Аа х аа - 50 % детей будет с патологической аллелью А. Если пенетрантность данного признака составляет 0,25, то фенотипически признак проявится только у 12,5 % детей Цитогенетические методы - группа методов, основанных на микроскопическом изучении хромосом человека. Эти методы применяются при диагностике хромосомных болезней. Наиболее распространенный из них - кариотипирование - рутинный метод изучения кариотипа, позволяет выявить нарушения в числе и структуре хромосом. С помощьюметода дифференциального окрашивания можно изучить характер хромосомных аберраций и их локализацию по характерному окрашиванию по длине хромосомы. В основном, хромосомы изучаются на стадии метафазы митоза. Для более точного анализа хромосомы можно изучать на стадии прометафазы, когда они еще не полностью спирализованы и по длине хромосомы можно различить не только районы и сегменты, но и субсегменты. Это позволяет обнаружить микроперестройки хромосом. 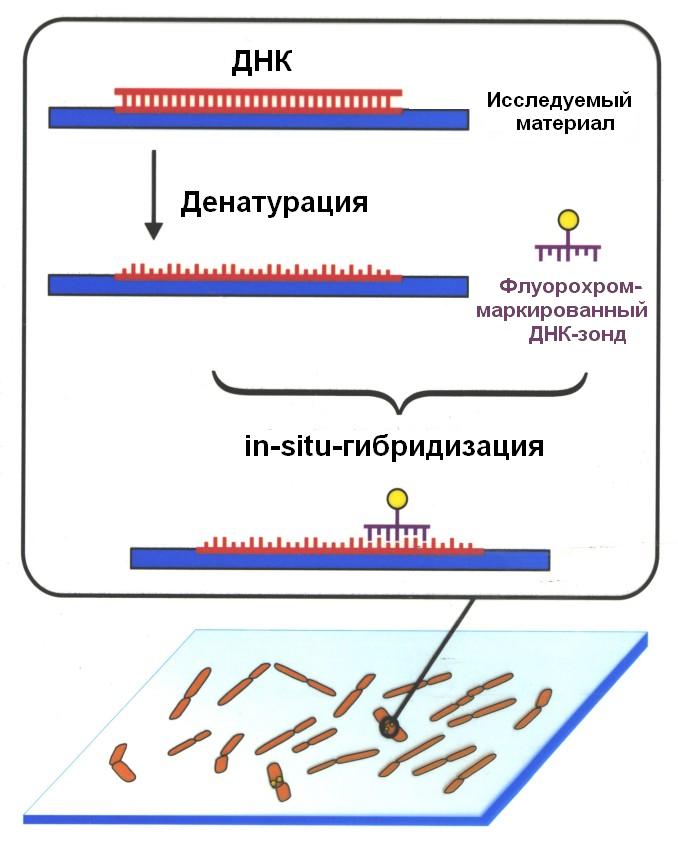 Рис.2 Метод флуоресцентной гибридизации. Благодаря успехам молекулярной генетики человека, разработан принципиально новый метод изучения хромосом - метод флуоресцентной гибридизации (FISH-метод). Для этого применяются ДНК-зонды – одноцепочечные фрагменты ДНК с известным нуклеотидным составом (копия нужного гена), которые комплементарно связываются с соответствующим участком хромосомы. Если такой ДНК-зонд пометить флуоресцентной меткой, то под микроскопом светящаяся точка будет указывать место локализации данного зонда на хромосоме. С помощью этого метода можно локализовать ген, расшифровать сложные перестройки между несколькими хромосомами, что недоступно для других цитогенетических методов (Рис.2). Применяется для диагностики хромосомных болезней, связанных со сложными изменениями структуры хромосом. Экспресс-методы - это быстрые, дешевые и легкие в исполнении предварительные методы диагностики. При этом используются легкодоступные материалы (кровь, моча) в малых количествах. Экспресс-выявление Х- и У-хроматина позволяет определить пол и изменения в числе половых хромосом. Чаще осуществляется посредством соскоба клеток слизистой оболочки щеки (буккальный эпителий) и изучения окрашенных препаратов под микроскопом. Этот метод позволяет определить количество Х-хромосом в кариотипе по количеству телец Барра (их на одну больше, чем количество Х-хромосом). Тельце Барра представляет собой инактивированную одну из Х-хромосом, и обнаруживается в 5-60 % клеток нормальных женщин и 2-5 % клеток мужчин в виде овального тельца под ядерной мембраной (Рис.3,а). Для выявления У-хроматина мазки окрашивают 0,005 % раствором акрихин-иприта и просматривают в люминесцентный микроскоп. При этом У-хромосома дает яркое зеленое свечение, что позволяет установить количество У-хромосом в кариотипе (Рис.3,в,г). В мазках крови можно определять половой Х-хроматин путем подсчета нейтрофильных клеток, где инактивированная Х-хромосома выглядит в виде выроста ядра в форме барабанной палочки (метод барабанных палочек) (Рис.3,б). 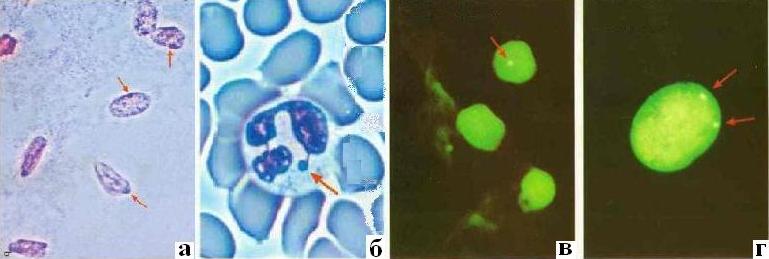 Рис.3 Цитогенетические экспресс методы выявления полового хроматина (а- тельца Барра (указано стрелкой) в соматических клетках; б- барабанные палочки в нейтрофилах крови; в,г- половой У-хроматин при люминесцентной микроскопии). Молекулярно-генетические методы - это большая и разнообразная группа методов, позволяющих выявить различные вариации в структуре ДНК, вплоть до расшифровки первичной последовательности нуклеотидов. Основные этапы и варианты молекулярно-генетических методов:
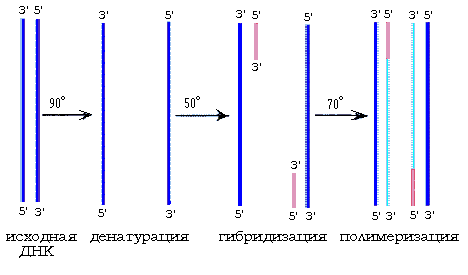 Рис. 4 Стадии одного цикла ПЦР
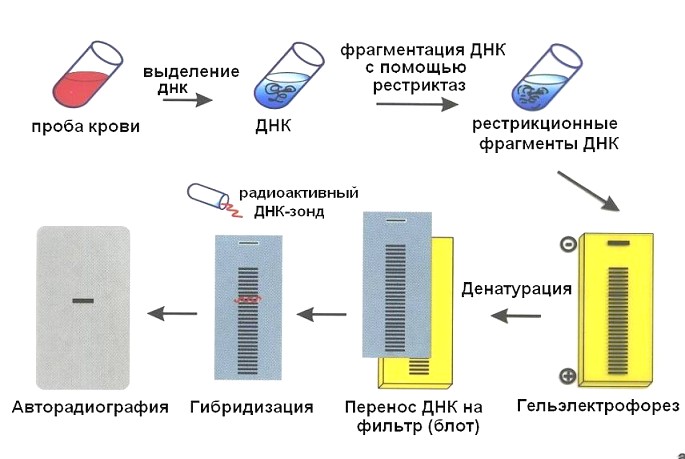 Рис.6 Выделение фрагмента ДНК с помощью блот-гибридизации по Саузерну.
Благодаря разработке молекулярно-генетических методов в настоящее время стало возможным диагностировать практически любую болезнь, связанную с нарушением в структуре генетического материала. Эти технологии нашли широкое применение в связи с исследованиями генома человека. Большинство методик автоматизированы, и в будущем, могут стать рутинными методами расшифровки генома каждого человека. Биохимические методы - это группа методов, позволяющих выявить дефект обмена какого-то вещества, путем определения его в крови, моче и других жидких средах организма. С их помощью описано более 1000 врожденных аномалий обмена веществ, выражающихся в виде накопления или наоборот дефицита каких-то продуктов. Особо значима роль биохимических методов в выявлении гетерозиготного носительства патологического гена (метод нагрузки). Эти методы применяются также в скрининговых (просеивающих) программах при широкомасштабных исследованиях населения для первичного выявления людей с некоторыми наиболее распространенными заболеваниями. Биохимические экспресс-методы – легкие и быстрые методы диагностики некоторых болезней обмена веществ. Например, добавление хлорного железа в мочу новорожденных вызывает позеленение окраски - что является признаком наличия фенилпировиноградной кислоты в моче. Микробиологический ингибиторный тест Гатри – изначально был предложен для диагностики биохимических нарушений у новорожденных. Из пятки новорожденного берут капли крови на диски фильтровальной бумаги и помещают их на культуру микроорганизмов, выращиваемых в определенной питательной среде. Ускорение роста этих микроорганизмов позволяет диагностировать наличие в крови младенца определенных аминокислот и углеводов выше нормы. Практическое занятие № 3 6.1. Тема: Хромосомные болезни. Аутосомные синдромы Информационно-дидактический блок: Хромосомными болезнями называются наследственные болезни человека, обусловленные изменениями числа или структуры хромосом. В медицинской генетике хромосомные болезни называются также хромосомными синдромами. Популяционная частота хромосомных синдромов составляет 5-7 на 1000 рождений. Хромосомные аномалии чаще всего наблюдаются при спонтанных абортах - до 60%, у мертворожденных - до 70% и у живорожденных - около 1%. Клинические и цитогенетические исследования, проводимые у новорожденных с хромосомной патологией, показывают, что жизнеспособность зависит от типа хромосомного нарушения. Большинство новорожденных с аутосомными трисомиями погибают в первые дни жизни. В свою очередь, у больных с аномалиями половых хромосом жизнеспособность снижена незначительно. Среди хромосомных нарушений принято выделять геномные и хромосомные нарушения. У человека найдены все формы хромосомных и геномных мутаций. К геномным мутациям относятся аномалии, характеризующиеся увеличением полного набора хромосом (полиплоидии) или изменением количества хромосом по одной из пар (анеуплоидии). У человека описано два вида полиплоидий – триплоидии и тетраплоидии. Полиплоидии не совместимые с жизнью и встречаются у абортусов и мертворожденных. Существует два основных механизма возникновения анеуплоидий: 1) неправильное расхождение гомологичных хромосом в мейозе, в результате в одну половую клетку попадают две хромосомы (это проводит к возникновению трисомной зиготы), а другую ни одной (зигота окажется моносомной); 2) неправильное расхождение гомолигичных хромосом в процессе коньюгации. К нарушению конъюгации хромосом могут приводить некоторые структурные хромосомные перестройки. Обусловливают различные изменение структуры хромосом. Хромосомные мутации являются причиной гибели около 30% зигот, 50% случаев самопроизвольных абортов и около 5% мертворождений. Большая часть геномных и хромосомных мутаций, приводящих к развитию хромосомных синдромов, элиминируется во внутриутробном периоде. У человека описано около 100 хромосомных синдромов. В практической медицине хромосомные синдромы можно распознать по следующим фенотипическим (клиническом) признакам: 1) низкая масса при рождении; 2) множественные пороки развития; 3) отставание в умственном и физическом развитии; 4) нарушение полового развития. К хромосомным болезням относят группу врожденных патологий, которые возникают в результате нарушения числа и структуры хромосом в соматических и половых клетках человека. Аутосомные синдромы Об общей характеристике аутосомных синдромов, следует помнить, что все моносомии по любой из аутосом обычно приводят к внутриутробной гибели плода. Чаще всего в материалах спонтанных абортусов встречаются моносомии. При трисомиях аутосом летальность гораздо меньше, однако родившиеся дети имеют тяжелейшие врожденные пороки развития. Наиболее частой причиной трисомии является нерасхождение хромосом в гаметогенезе родителей. Нерасхождение хромосом в мейозе резко возрастает с возрастом матери, при этом возраст отца практически не имеет значения. Это связано с особенностями мейоза у женщин. Наиболее благоприятное положение наблюдается при наличии в организме мозаицизма. Дети с мозаичным кариотипом обладают повышенной жизнеспособностью, а клиническая картина у них менее выражена. Известно, что среди живорожденных с аутосомными синдромами чаще всего встречаются полные трисомии по 13, 18 и 21 хромосомам, среди которых 75% приходится на долю синдрома Дауна. Остальные аутосомные трисомии являются еще более редкими, и их носители погибают в раннем неонатальном возрасте. Числовые аномалии хромосом Трисомия по 21 хромосоме (синдром Дауна) Синдром Дауна встречается в популяциях со средней частотой 1:700 живорожденных детей и увеличивается с возрастом матери. У женщин старше 45 лет частота рождения больных детей достигает 4% . Цитогенетический анализ больных детей выявляет в 95 % случаев наличие добавочной 21 хромосомы (кариотип 47,21+). В 3 % случаев имеет место транслокация 21 хромосомы на одну из хромосом группы Д, чаще всего 13. В 2 % случаев выявляется мозаицизм: часть клеток больного имеют в генотипе трисомию,часть-нормальный, двойной набор 21 хромосомы. Синдром Дауна характеризуется умственной отсталостью, брахицефалией, уплощением затылка и лица, коротким носом, аномалией прикуса, аномалиями строения скелета (короткие пальцы кисти и стоп), наличием глубокой поперечной складки на ладони (в общей популяции встречается в 1 %, у больных – в 40 %). Все дети при этой аномалии имеют характерный фенотип и очень похожи друг на друга. При патологоанатомическом исследовании мозг больных уменьшен в размерах и массе, ствол мозга и мозжечок маленькие, борозды и извилины развиты не полностью. У больных с синдромом Дауна наблюдаются множественные пороки развития сердечно-сосудистой системы (дефект межжелудочковой перегородки, незаращение боталлова протока), пороки развития почек, мочевых путей, желудочно-кишечного тракта, нарушение слуха. У этих больных чаще возникают инфекционные и злокачественные заболевания, что связано с дефектностью иммунной системы. Дети с синдромом (трисомная форма) чаще рождаются у женщин старше 35 лет. Причины такой зависмости на сегодня до конца не ясны. Трисомная форма синдрома Дауна практически не наследуется, повторный риск оценивается в 1%. Лечение при этом заболевании малоэффективно, в основном симптоматическое (витамины, гормональные препараты, занятия с логопедом, массаж и т.д.) Трисомия по 13 хромосоме (синдром Патау) Популяционная частота синдрома Патау составляет 6000 - 1:10000 рождений (кариотип 47, 13+). Большинство больных детей умирает в первые недели жизни. Цитогенетически у больных выявляется трисомия по 13 хромосоме (кариотип 47, 13+), в редких случаях выявляется робертсоновкая транслокация 13 хромосомы с одной из хромосом группы Д(14-15 хромосомы). Повторный риск при трисомии по 13 хромосоме низкий, при транслокационной форме –около 1 %. Синдром Патау - при данной болезни наблюдаются дефекты внутренних органов (аномалии конечностей, волчья пасть, микроцефалия, глухота, анофтальмия), которые резко снижают жизнеспособность. Новорожденные с синдромом Патау погибают сразу после рождения или в первые месяцы жизни. При патологоанатомическом исследовании наблюдаются множественные пороки развития всех органов и систем. Масса мозга уменьшена, часто отсутствует передний мозг, мозжечок недоразвит, иногда мозг не разделен на полушария. Сердечно-сосудистая система: дефекты межжелудочковой и межпредсердной перегородок, камеры сердца расширены; в легких: явления хронической неспецифической пневмонии; отмечаются аномалии почек: гидронефроз, кистозная почка, удвоение мочеточника в среднем в 61,5 %. Прогноз неблагоприятный, успешных методов лечения нет. Трисомия по 18 хромосоме (синдром Эдвардса) Популяционная частота составляет 1:5000 - 7000 рождений; больные девочки рождаются в 3 раза чаще мальчиков. Цитогенетический анализ выявляет в 90% случаев трисомию по 18 хромосоме (кариотип 47, 18+), в 10% случаев имеет место мозаицизм или робертсоновская транслокация 18 хромосомы с другими хромосомами. Генетический риск низкий. Синдром Эдвардса - при данном синдроме наблюдается микроцефалия, косоглазие, пупочная грыжа, синдактилия. Очень часто характерное расположение пальцев (65 %), ноги узкие и выпуклые, вывих бедра (30 %), конская стопа с внутренней косолапостью, ограниченная подвижность бедра, узкий таз. Аномалии внутренних органов: пороки сердца (дефекты межжелудочковых и межпредсердных перегородок), гетеротрофия в мозжечке, крипторхизм, эктопия или подковообразная почка, гидронефроз, удвоение мочеточников. Другие признаки: тяжелая задержка умственного развития, частые инфекции, гипертонус, лица женского пола поражаются чаще, чем мужского (1:3). Прогноз жизни при синдроме Эдвардса не благоприятен. 30 % детей погибают в первом месяце, 50 % во втором и менее 10 % доживают до года, успешных методов лечения нет. |