зайко. Н. Н. Зайко Патологическая физиология Введение Предмет и задачи патологической физиологии Патологическая физиология есть наука, изучающая жизнедеятельность больного организма. Программа
 Скачать 7.32 Mb. Скачать 7.32 Mb.
|

|
Причиной жирового перерождения печени может быть любое нарушение, которое дезинтегрирует обмен и синтез липидов в печени: 1. усиленный печеночный липогенез; 2. снижение окисления жирных кислот; 3. повышенный липолиз жировой ткани; 4. замедление выделения липопротеидов очень низкой плотности (ЛПОНП). Продукция ЛПОНП в печени требует сочетания процессов липидного и белкового синтеза и их нарушение приводит к аккумуляции жира в печени. Недостаточное питание и дефицит аминокислот нарушают синтез аполипопротеидов и снижают выработку липопротеидов. К такому же результату приводит усиленный липолиз в жировой ткани при голодании или сахарном диабете, когда нарушается включение липидных и белковых предшественников в обмен липопротеидов. В патогенезе жировой инфильтрации большое значение имеет нарушение образования фосфолипидов. Достаточное содержание их в печени обеспечивает тонкое диспергирование жира и возможность удаления его из печени. Фосфолипиды входят в состав ?-липопротеидов и облегчают их выход из клеток печени. Часть жирных кислот участвует в образовании фосфолипидов и в их составе покидает печень. Кроме того, в молекуле фосфолипидов жирные кислоты лучше окисляются. Необходимыми компонентами основного фосфолипида печени – лецитина – являются холин и метионин, который дает свои метильные группы для образования холина. Поэтому недостаток в пище холина, метионина и других липотропных веществ (инозит, нуклеиновые кислоты) приводит к развитию алипотропной жировой инфильтрации печени. К такому же результату приводит дефицит эндогенного липотропного фактора -липокаина, который образуется в эпителии мелких протоков поджелудочной железы. Липокаин активизирует образование фосфолипидов в печени, окисление в ней жирных кислот и предохраняет печень от ожирения. Недостаточность этого фактора играет важную роль в патогенезе ожирения печени при сахарном диабете. Нарушение образования холина возможно при дефиците витамина В, фолиевой и пантотеновой кислоты. Нарушения промежуточного жирового обмена Одним из наиболее важных нарушений промежуточного обмена жира является усиление кетогенеза. Образующиеся в процессе ?-окисления жирных кислот кетоновые тела занимают одно из центральных мест в системе обеспечения организма энергией, конкурируя в этом отношении с глюкозой. При невозможности использовать в качестве источника энергии глюкозу в организме усиливается липолиз и кетогенез. Такой кетоз может наблюдаться и в физиологических условиях (физическая работа, эмоциональное напряжение, поздние сроки беременности), но тогда он не бывает продолжительным, уровень кетоновых тел в крови не превышает 0,1 мМ, поскольку происходит быстрая утилизация в качестве энергетического сырья (физиологический кетоз). При патологическом кетозе производство кетоновых тел превышает утилизацию. Обычно это бывает при усилении липолиза в жировой ткани, когда печень не использует всех жирных кислот для синтеза триглицеридов и часть их включается в процесс ?-окисления и кетогенеза. Таково происхождение кетоза при голодании, сахарном диабете. При значительном накоплении кетоновых тел в крови (свыше 0,1, а иногда до 20 мМ) возникает угрожающий жизни метаболический ацидоз (см. "Диабетическая кома"). Нарушения обмена жира в жировой ткани Жировая ткань характеризуется интенсивным метаболизмом, обильным кровоснабжением и является своего рода саморегенерирующимся энергетическим аккумулятором. Накопление энергии в виде нейтральных жиров происходит в ней после каждого приема пищи, а мобилизация энергии – в любое время под влиянием импульсов, освобождающих жирные кислоты. Если в течение длительного времени накопление превышает расход энергии – появляетсяожирение. В жировых клетках функционируют все три метаболических пути: гликолиз, пентозный цикл и цикл Кребса, в них осуществляется синтез жирных кислот, липогенез и липолиз. На рис. 14.7 показана роль нервной системы и гормонов в депонировании жира и липолизе. Липолизактивируется адреналином, кортикотропином и глюкагоном посредством циклической АМФ, активирующей триглицеридную липазу. Из гормональных факторов, обладающих жиромобилизующим эффектом, следует отметить соматотропин, тиро тропин и тироксин. Известно, что в период усиленного роста, а также при гипертиреозе наступает значительное похудание. Стимулирующее липолиз действие оказывает также выделенный из аденогипофиза ?-липотропин. 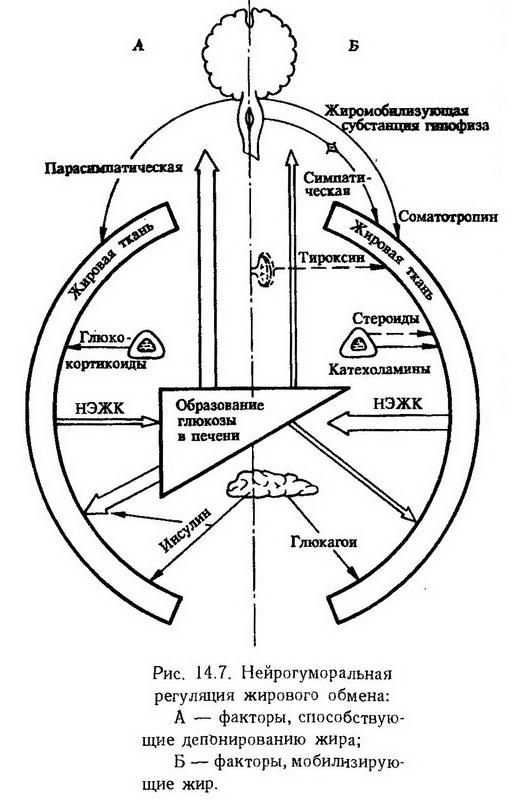 Гликокортикоиды способствуют усилению мобилизации жира из жировой ткани и тормозят липогенез. Но это действие в организме может перекрываться другими эффектами данных гормонов: способностью вызывать гипергликемию и стимулировать секрецию инсулина, накопление гликогена в печени, что приводит к торможению мобилизации жира и его отложению в жировой ткани; способностью в больших дозах задерживать жиромобилизующее и стимулирующее окисление жиров действие соматотропина. Этим можно объяснить накопление жира в жировых депо при гипергликокортицизме (болезни и синдроме Иценко-Кушинга). Кроме того, при этом состоянии увеличено образование дигидрокортизона, который стимулирует пентозный цикл и превращение углеводов в жиры (С. М. Лейтес). Кортикотропин, стимулируя секрецию гликокортикоидов, может влиять на жировой обмен в том же направлении, но, помимо этого, обладает еще и экстраадреналовым жиромобилизующим действием. Липогенез, т. е. анаболические процессы в адипоцитах, регулируется инсулином. Инсулин стимулирует синтез нейтральных жиров из глюкозы и жирных кислот, тормозит липолиз, снижая уровень сахара в крови, повышает аппетит. На рис. 14.8 показана роль инсулина и адреналина в метаболизме адипоцита и соотношении процессов липогенеза и липолиза. 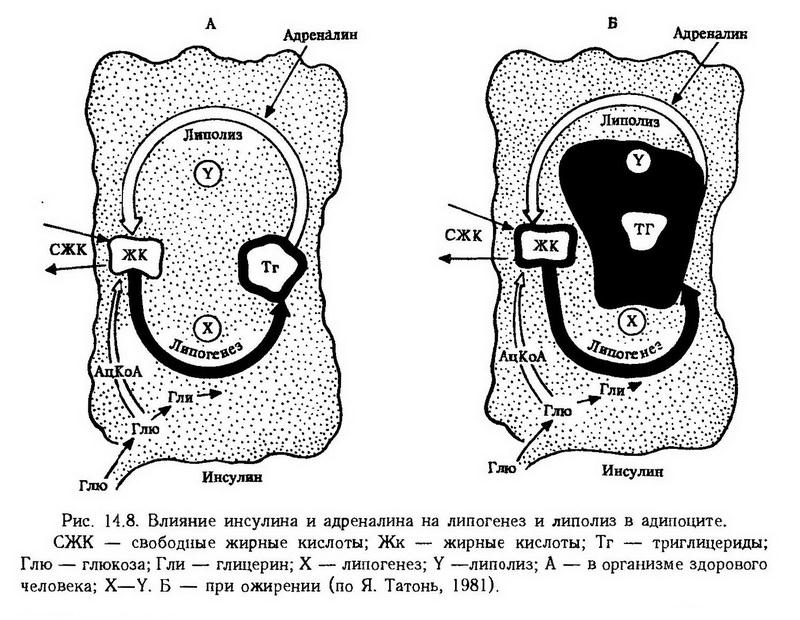 Гормоны влияют на метаболизм адипоцитов через посредство специфических рецепторов. Дефицит инсулиновых рецепторов может быть причиной резистентности тканей к инсулину и индуцировать гиперинсулинизм. При ожирении наблюдается не только гиперинсулинизм, но и снижение секреции глюкагона, соматотропина, повышение секреции кортизола и снижение реакции жировой ткани на катехоламины. Роль нервной системы в регуляции жирового обмена подтверждается данными о том, что длительное эмоциональное напряжение приводит к мобилизации жира из жировых депо и похуданию. Такой же эффект наблюдается при раздражении симпатических нервов. Десимпатизация препятствует выходу жира из депо. Раздражение парасимпатических нервов сопровождается отложением жира. Синтез и распад триглицеридов регулируется также уровнем глюкозы в крови. При избытке глюкозы часть НЭЖК, как второй источник энергии, изымается из обращения и откладывается в жировых депо; при дефиците глюкозы из депо мобилизуются жиры. Этот процесс саморегуляции является одним из звеньев в сложной системе регуляции жирового обмена, осуществляемого нервной и эндокринной системами. Ожирение – избыточное отложение жира в жировой ткани. Среди взрослого населения экономически развитых стран у 30 – 60% масса тела превышает норму на 20% и более. Ожирение чаще встречается у женщин и в возрастных группах старше 50 лет. Этиология. Ожирение является результатом расстройства гомеостаза энергетического обмена. В его возникновении принимают участие внутренние и внешние факторы, которые меняют поведение человека в отношении питания. Факторы, регулирующие поведение человека в отношении потребления пищи, определяются генетически-конституциональными особенностями индивидуума, а также влияниями внешней среды. Среди последних определенную роль играет питание матери в предродовой период; ребенка – в грудном возрасте и раннем детстве; типы безусловных рефлексов, связанных с питанием; семейные, национальные традиции; уровень материальной обеспеченности и доступность пиши; состояние двигательной активности. Повышенное потребление пищи является одной из основных причин ожирения. Реже причиной ожирения бывают первичные нарушения нервно-гормональной регуляции, изменения в обмене адипоцитов или генетические факторы. По этиологии выделяют ожирение первичное (конституциональное) – 55 – 65% и вторичное (симптоматическое), оно подразделяется на гормональное (около 20%) и церебральное (16 – 20%). Несомненна роль наследственности в ожирении. Наследоваться могут структура и функция систем, регулирующих алиментарное поведение, особенности метаболизма адипоцитов и миоцитов. Имеются наблюдения о том, что ожирение развивается в нескольких поколениях одной и той же семьи. Отмечена высокая конкордантность по этому признаку у однояйцовых близнецов. Однако эти данные не являются прямыми доказательствами роли наследственности в ожирении, поскольку здесь нельзя исключить влияние окружающей среды, привычек, касающихся видов пищи, а также образа жизни. Более убедительные данные получены в эксперименте. Экспериментальное ожирение. Имеется несколько линий мышей, у которых ожирение проявляется как генетически обусловленное нарушение, обычно передающееся аутосомно-рецессивно. У мышей с наследственным ожирением отмечаются гипергликемия, гиперлипемия, усиление липогенеза, гиперинсулинемия. У них обнаружены повышение чувствительности к гипергликемическому действию гормона роста и глюкагона, устойчивость к инсулину, снижение числа инсулиновых рецепторов в адипоцитах. Ферментативные изменения в гепатоцитах указывают на увеличенное использование фосфатоглицерола для липогенеза. Описан штамм желтых мышей, у которых сопряжены два свойства: желтая окраска шерсти и предрасположение к ожирению. Мыши характеризуются повышенным потреблением углеводов, резистентностью к инсулину при нормальной гликемии. У другой линии мышей ожирение и гипергликемия, появляющиеся на втором месяце жизни, сочетаются с гиперхолестеринемией, повышением чувствительности к действию гормона роста и глюкагона. У этих животных обнаруживается гипертрофия островков поджелудочной железы и гиперинсулинемия. Описана наследственная форма ожирения у мышей, сочетающаяся с аденомой гипофиза и гипертрофией панкреатических островков. Нарушение соотношения между приходом и расходом энергии может быть вызвано воздействием на гипоталамические центры, с которыми связано ощущение голода и сытости. Электролитическое разрушение вентромедиальных ядер – "центра сытости", угнетающе действующего на "пищевой центр", у крыс, кошек и обезьян вызывает гиперфагию с последующим развитием гипоталамического ожирения. Такой же эффект вызывает ауротиоглюкоза, избирательно поражающая вентромедиальные ядра. Экспериментальное гипоталамическое ожирение является аналогом диэнцефального ожирения у людей, которое может развиться в результате перенесенного энцефалита, менингита, травмы головного мозга. Гормональное ожирение в эксперименте развивается после удаления щитовидной железы, а также после введения инсулина с глюкозой. Ожирение после кастрации связано с развивающейся в результате компенсации гормональных нарушений гипертрофией коры надпочечников и гиперинсулинизмом. При этом активируются процессы глюконеогенеза и перехода углеводов в жир. Возможно, такое же происхождение имеет ожирение у женщин в период климакса. Экзогенное ожирение моделируется ограничением двигательной активности животных и перекармливанием, особенно углеводной пищей. Патогенез. В развитии ожирения имеют значение три основных патогенетических фактора: повышенное поступление пищи, несоответствующее энергетическим затратам; недостаточная мобилизация жира из депо; избыточное образование жира из углеводов (С. М. Лейтес). Избыточное потребление пищи, вызванное усилением аппетита, может быть обусловлено повышенной возбудимостью "пищевого центра", расположенного в переднебоковых ядрах задней гипоталамической области. Изменения, на которые реагирует пищевой центр, могут быть причиной длительного пищевого возбуждения, и вследствие этого – алиментарного ожирения: Так, все состояния, которые стойко понижают уровень глюкозы в крови, например, некоторое повышение функции панкреатических островков, сопровождаются чувством голода, которое обусловливает возможность переедания. В деятельности пищевого центра имеет также значение сигнализация, Поступающая с рецепторов пищевого канала. Определенная степень растяжения желудка тормозит деятельность пищевого центра. При понижении чувствительности нервных окончаний в стенке желудка торможение центра развивается только при чрезмерном растяжении желудка, что также создает предпосылки к перееданию и ожирению. Нарушение баланса энергии возможно и при переходе от физического труда к образу жизни, не требующему большой физической нагрузки, если прежняя степень возбудимости пищевого центра сохранена. При нормальной функции пищевого центра ожирение может быть связано с нарушением мобилизации жира из жировых депо в качестве источника энергии. Как было сказано выше, регуляция процесса мобилизации и отложения жира осуществляется нервной и эндокринной системами. Так, снижение тонуса симпатической нервной системы может вызвать задержку мобилизации и выхода жира из жировой ткани. У кошек при односторонней перерезке чревного нерва в случае голодания количество жира в околопочечной клетчатке больше на денервированной стороне. При болезни Барракера-Симонса – прогрессивной липодистрофии, связанной с поражением центров промежуточного, спинного мозга и узлов симпатического ствола, – наблюдается исчезновение жира из жировой ткани на голове и грудной клетке с одновременным отложением его в нижней половине тела. Нарушение мобилизующего жир влияния гормонов наблюдается при патологии гипофиза, щитовидной железы, надпочечных и половых желез. У людей может наблюдаться ожирение, характеризующееся гиперинсулинизмом, резистентностью к инсулину и гипергликемией. Считают, что основные повреждения при этом находятся на уровне клеток-мишеней. Они связаны с уменьшением числа рецепторов для инсулина, обусловливающим резистентность к инсулину и компенсаторный гиперинсулинизм. В настоящее время в патогенезе ожирения учитываются особенности и самой жировой ткани, число и величина жировых клеток – адипоцитов. Количество жировых клеток – фактор генетически обусловленный, а величина их зависит от возраста, пола, воздействия регуляторных и метаболических факторов. Число жировых клеток относительно постоянно и с возрастом не меняется. У женщин оно больше, чем у мужчин. У молодых людей число жировых клеток составляет 3 • 1010, содержание жира в адипоците – 0,6 мкг, общее количество жира в организме – примерно 18 кг. У лиц с небольшой физической активностью и ожирением эти величины соответственно составляют: 4,6 • 1010; 1,1 мкг и 50 мг. Встречаются случаи ожирения, когда общее количество жира составляет более 70 кг, при нормальном числе адипоцитов, однако масса одной клетки равна 1,6 мкг. В других случаях масса адипоцитов остается нормальной, а их число достигает 9 • 1010. Общее количество жира может составлять 100 кг и более. Патогенетическая классификация ожирения, основанная на критерии величины и количества адипоцитов, выделяет два типа ожирения: гипертрофическое игиперпластическое. Гипертрофическое ожирение зависит от количества жира в каждом адипоците, что взаимосвязано с повышенной концентрацией инсулина, гиперлипемией, снижением толерантности к глюкозе. Нередко эта форма ожирения осложняется развитием в молодом возрасте атеросклероза и диабета. Гиперпластическое ожирение связано с увеличением количества адипоцитов, которое зависит от генетических факторов или влияний, регулирующих морфогенез жировой ткани в эмбриональном периоде и раннем детстве. Ожирение неблагоприятно отражается на жизнедеятельности организма. В молодом возрасте, когда адаптационные возможности выражены лучше, отрицательное действие ожирения проявляется в меньшей степени, а с возрастом количество осложнений, связанных с ожирением, увеличивается. Смертность у страдающих ожирением в возрасте 20 – 24 лет на 3% выше, чем у лиц с нормальной массой тела, у лиц в возрасте 40 – 55 лет она выше на 50%. В связи с отложением большого количества жира и увеличением нагрузки на большинство жизненно важных органов далеко зашедшее ожирение вызывает ряд функциональных изменений в них, а также нарушения метаболизма. Прежде всего нарушается обмен в жировой ткани, где повышается скорость синтеза триглицеридов и липопротеидов, нарушается способность к мобилизации жировых резервов, наблюдается гиперлипемия, повышение уровня свободных жирных кислот, гиперхолестеринемия. Нарушения в углеводном обмене выражаются в ограничении обмена глюкозы, повышении содержания гликогена в печени. В мышечной ткани нарушается утилизация глюкозы, несмотря на гиперинсулинизм. Дыхательный коэффициент, равный 0,7 – 0,74, свидетельствует о том, что в качестве источника энергии используются в основном жирные кислоты. Отложение жира в миокарде значительно снижает сократительную функцию сердца. Ожирение зачастую сопровождается атеросклерозом, повышением артериального давления, свертываемости крови, развитием тромбоза. При этом ухудшается легочная вентиляция, уменьшается жизненная емкость легких, появляется склонность к застою крови и развитию в дыхательных путях хронического воспаления. Одышка возникает даже при небольшой физической нагрузке. Появляется циркуляторная и дыхательная гипоксия. Сочетание ожирения с сахарным диабетом возникает в случае инсулинрезистентности, связанной с уменьшением числа рецепторов к инсулину на поверхности жировых клеток. Компенсаторная гипертрофия и гиперплазия панкреатических островков, обеспечивая повышенную секрецию инсулина (гиперинсулинизм) для преодоления резистентности, со временем сменяется истощением. В этом случае полагают, что ожирение является этиологическим фактором сахарного диабета. Наследственные нарушения жирового обмена Известно несколько наследственных нарушений жирового, липоидного обменов, которые связаны между собой. К редко встречающимся относится эссенциальная гиперлипемия. Она обусловлена гиперлипопротеидемией III типа, которая характеризуется наличием в плазме ненормальных липопротеидов очень низкой плотности, содержащих особенно в большом количестве триглицериды. Предполагается, что генетический дефект приводит к блокаде поздних стадий катаболизма липопротеидов очень низкой плотности. Наследуется как признак неполной доминантности, сочетается с ожирением. Гиперлипопротеидемия IV типа или семейная гипербетали-попротеидемия. Повышенное количество триглицеридов синтезируется в печени, эритроцитах. При этом отчетливо проявляется индукция синтеза жиров углеводами и тоже сопровождается ожирением. Среди наследственно обусловленных нарушений обмена холестерина наиболее распространенной является семейная гиперхолестеринемия. Она проявляется в виде ксантоматоза, атероматоза и развитием в молодом возрасте ишемической болезни сердца. В плазме крови повышается концентрация липопротеидов низкой плотности (ЛПНП). Наследование болезни аутосомно-доминантное; гомозиготы поражаются более тяжело, нередко инфаркт миокарда возникает в детском возрасте; генетический дефект – отсутствие на клеточных мембранах рецепторов для ЛПНП. Функция рецептора состоит в связывании ЛПНП и введении их в клетку, где они распадаются с освобождением свободного холестерина. Эта аномалия обмена предрасполагает к ишемической болезни сердца. Первые приступы стенокардии в большинстве случаев развиваются до 30-летнего возраста, ишемическая болезнь – к 50 годам, и половина страдающих этой болезнью умирают в возрасте до 60 лет. Липидозы относятся к болезням накопления, обусловленных дефектами специфических лизосомальных гидролаз. Болезнь Вольмана – редкое аутосомно-рецессивное заболевание, которое в первые недели жизни проявляется рвотой, диареей со стеатореей, гепатоспленомегалией и двусторонним кальцинозом надпочечных желез. Дети умирают в возрасте до 6 мес. Генетический дефект – отсутствие кислой липазы лизосом, обусловливающее накопление эфиров холестерина в лизосомах печени, селезенки, надпочечных желез, гемопоэтической системы и тонких кишок. Болезнь Шюллера – Кристиана характеризуется отложением в клетках грануляционной ткани, разрастающейся в костях и в большинстве внутренних органов, холестерина и его эфиров. Характерны при этом деструктивные изменения в костях. При некоторых патологических состояниях наблюдается избыточное отложение в тканях фосфолипидов. Все они наследуются по аутосомно-рецессивному типу. Так, приболезни Гоше в связи с отсутствием гликоцереброзидазы цереброзиды откладываются в макрофагальных клетках селезенки, печени, лимфатических узлов и костного мозга. Ведущими симптомами заболевания являются спленомегалия, увеличение печени, а также изменения в костях, проявляющиеся в виде остеопороза. При болезни Ниманна – Пика наблюдается отложение фосфатида сфингомиелина в клетках различных органов. Генетический дефект – дефицит сфингомиелиназы. Болезнь проявляется резким увеличением печени и селезенки, замедлением психического развития ребенка, появлением слепоты и глухоты. Чаще всего дети погибают в возрасте до 2 лет. Амавротическая семейная идиотия является результатом отложения ганглиозидов в клетках нервной системы, что сопровождается атрофией зрительных нервов, а также слабоумием. Нарушения белкового обмена Поскольку белки занимают центральное положение в осуществлении процессов жизнедеятельности организма, то и нарушения обмена белков в различных вариантах являются компонентами патогенеза всех без исключения патологических процессов. Для получения полного представления о нарушениях белкового обмена, исходят из понятия об азотистом равновесии. У нормального взрослого человека количество азотистых веществ, выводимых из организма, равняется тому, которое он получает с пищей. В растущем организме, при беременности, при введении или избыточной выработке гормонов анаболического действия, при откармливании после истощающих заболеваний азота выводится меньше, чем поступает, т. е. анаболические процессы преобладают над катаболическими (положительный азотистый баланс). Отрицательный азотистый баланс имеет место при потере белков или большом расходе их организмом. Это может быть при голодании, потере белков через почки (протеинурия), кожу (ожоги), кишки (понос), при тиреотоксикозе, инфекционной лихорадке. Нарушения белкового обмена возможны на всех этапах, начиная с всасывания и кончая выведением из организма конечных продуктов обмена. В такой последовательности эти нарушения будут рассмотрены ниже. Нарушения всасывания и синтеза белков Поскольку в организме практически нет депо белков, а источником аминокислот для их синтеза служат в основном компоненты пищи, то, естественно, при нарушении переваривания и всасывания белков развивается алиментарная белковая недостаточность. Наблюдается она при воспалительных и дистрофических изменениях различных отделов кишок, сопровождающихся нарушением их секреторной и моторной функций, при голодании, несбалансированном по аминокислотному составу пищи. Однако для нормального синтеза белков необходимо не только достаточное количество аминокислот, но и правильное и активное функционирование системы этого синтеза и кодирующих его генетических структур. Нарушение продукции белка может быть приобретенным и наследственным. Оно выражается в изменении количества синтезированных молекул или появлении молекул с измененной структурой. Увеличение или уменьшение количества синтезируемого белка чаще всего связано с изменением регуляторных влияний со стороны ряда гормонов, нервов и иммунной системы. Кроме того, к нарушению протеосинтеза может приводить конденсация хроматина при различных патологических процессах в клетках, нерегулируемая скорость списывания матричной РНК при нарушении функционирования гена – регулятора или оператора (в опухолевых клетках), а также дефекты в структуре рибосом, возникающие, например, под влиянием стрептомицина. Синтез белков с измененной структурой обычно бывает следствием ошибок в геноме. Это может проявляться нарушением аминокислотного состава белковой молекулы (например, молекула гемоглобина при серповидно-клеточной анемии), укорочением молекул (когда транскрипция информации с ДНК-матрицы идет только до дефекта в ней), а также синтезом аномально длинных белков, если мутация произошла в "стоп-сигнале" гена и терминирующий кодон исчез. Примером этого может служить появление удлиненных альфа-цепей гемоглобина. Продукция белков с измененной структурой может быть также следствием нарушения одного из звеньев белоксинтезирующей системы – аппарата трансляции либо посттрансляционной модификации молекул. С увеличением частоты ошибок трансляции в процессе жизни связывают старение организма. Нарушения обмена аминокислот Нарушение трансаминирования и окислительного дезаминирования. Процессы трансаминирования и дезаминирования имеют универсальное значение для всех живых организмов и всех аминокислот: трансаминирование приводит к образованию аминокислот, дезаминирование – к их разрушению. Сущность реакции трансаминирования заключается в обратимом переносе аминогруппы от аминокислоты на а-кетокислоту без промежуточного образования свободного аммиака. Реакция катализируется специфическими ферментами: аминотрансферазами или трансаминазами, кофакторами которых являются фосфорилированные формы пиридоксина (пиридоксальфосфат и пиридок-саминофосфат). Нарушения реакции трансаминирования могут возникать по нескольким причинам: это прежде всего недостаточность пиридоксина (беременность, подавление сульфаниламидными препаратами кишечной флоры, частично синтезирующей витамин, торможение синтеза пиридоксальфосфата во время лечения фтивазидом). Снижение активности трансаминаз происходит также при ограничении синтеза белков (голодание, тяжелые заболевания печени). Если в отдельных органах возникает некроз (инфаркт миокарда или легких, панкреатит, гепатит и др.), то вследствие разрушения клеток тканевые трансаминазы поступают в кровь и повышение их активности в крови при данной патологии является одним из диагностических тестов. В изменении скорости трансаминирования существенная роль принадлежит нарушению соотношения между субстратами реакции, а также гормонам, особенно гликокортикоидам и гормону щитовидной железы, оказывающим стимулирующее влияние на этот процесс. Угнетение окислительного дезаминирования, приводящее к накоплению неиспользованных аминокислот, может вызвать повышение концентрации аминокислот в крови -гипераминоацидемию. Следствием этого является усиленная экскреция аминокислот почками (аминоацидурия) и изменение соотношения отдельных аминокислот в крови, создающие неблагоприятные условия для синтеза белковых структур. Нарушение дезаминирования возникает при недостатке компонентов, прямо или косвенно участвующих в этой реакции (недостаток пиридоксина, рибофлавина, никотиновой кислоты; гипоксия; белковая недостаточность при голодании). Нарушения декарбоксилирования. Являясь очень важным, хотя и не универсальным, направлением белкового обмена, декарбоксилирование протекает с образованием CO2 и биогенных аминов. Декарбоксилированию подвергаются только некоторые аминокислоты: гистидин – с образованием гистамина, тирозин – тирамина, 1-глутаминовая кислота – ?-аминомасляной кислоты, 5-гидрокситриптофан -серотонина, производные тирозина (3,4-диоксифенилаланин) и цистина (1-цистеиновая кислота) – соответственно 3,4-диоксифенилэтиламина (дофамин) и таурина. Биогенные амины, как известно, обладают специфической биологической активностью и увеличение их количества может вызвать ряд патологических явлений в организме. Причиной такого увеличения может быть не только усиление декарбоксилирования соответствующих аминокислот, но и угнетение окисления аминов и нарушение их связывания белками. Так, например, при гипоксических состояниях, ишемии и деструкции тканей (травмы, облучение и др.) ослабляются окислительные процессы, что способствует усилению декарбоксилирования. Появление большого количества биогенных аминов в тканях (особенно гистамина и серотонина) может вызвать значительное нарушение местного кровообращения, повышение проницаемости сосудов и повреждение нервного аппарата. Наследственные нарушения обмена некоторых аминокислот. Прохождение аминокислот через определенные метаболические пути детерминируется наличием и активностью соответствующих ферментов. Наследственное нарушение синтеза ферментов приводит к тому, что соответствующая аминокислота не включается в метаболизм, а накапливается в организме и появляется в биологических средах: моче, кале, поте, цереброспинальной жидкости. Клиническая картина такого заболевания определяется, во-первых, появлением слишком большого количества вещества, которое должно было метаболизироваться при участии заблокированного фермента, а во-вторых, дефицитом вещества, которое должно было образоваться. Таких генетически обусловленных нарушений обмена аминокислот известно довольно много; все они наследуются рецессивно. Некоторые из них представлены в табл. 4. Таблица 4. Наследственные нарушения аминокислот, связанные с отсутствием или низкой активностью ферментов
|