зайко. Н. Н. Зайко Патологическая физиология Введение Предмет и задачи патологической физиологии Патологическая физиология есть наука, изучающая жизнедеятельность больного организма. Программа
 Скачать 7.32 Mb. Скачать 7.32 Mb.
|

|
Глава 14. Типические нарушения обмена веществ. Нарушения регуляции обмена веществ Обмен веществ, или метаболизм, в организме определяется наследственными факторами и регулируется деятельностью эндокринной и нервной систем. В соответствии с этим и нарушения обмена веществ могут носить наследственный характер или возникать в результате нарушения функции регулирующих систем. Нарушения метаболизма могут проявляться на всех уровнях биологической организации – от молекулярного и клеточного до организменного. На клеточном уровне они связаны прежде всего с нарушением механизмов саморегуляции. Ведущая роль в осуществлении внутриклеточной саморегуляции принадлежит генетической информации. Большинство наследственных дефектов обмена веществ обусловлено мутацией генов, кодирующих синтез ферментов (наследственные ферментопатии). Значительно меньший удельный вес в развитии наследственной патологии имеют мутационные изменения структурных и транспортных белков. Сущность ферментопатии заключается в том, что при этом ферментный белок не синтезируется или синтезируется с измененной структурой, что изменяет его активность. При снижении активности фермента возможно накопление неметаболизирующегося субстрата или выпадение промежуточного продукта обмена. Повышение ферментативной активности обычно приводит к накоплению конечных продуктов обмена. Для координации метаболических реакций в клетке необходим постоянный приток информации, которую осуществляют медиаторы нервной системы и гормоны. Для восприятия информации клетки располагают специфическими рецепторами на поверхностной мембране, в цитоплазме либо в ядре. Патология рецепторов может служить одним из механизмов развития патологических процессов, например, при сахарном и несахарном диабете. Наряду с внутриклеточными механизмами саморегуляции организм располагает и более сложными – это нервно-гормональные механизмы регуляции. Гормональная регуляция на клеточном уровне может осуществляться также с помощью генетического аппарата путем индукции образования ферментов (например, инсулин индуцирует синтез ферментов гликолиза) или изменения активности имеющихся ферментов (адреналин активирует фосфорилазу, инсулин – гексокиназу). Нервная система осуществляет свою трофическую функцию, т. е. контролирует тканевый обмен, с помощью медиаторов, а также посредством аксоплазматического тока. При нарушении этой функции развивается нейродистрофический процесс. Нарушения обмена веществ на более высоком уровне биологической организации – органном и организменном – в большей степени зависят от состояния нейроэндокринной регуляции. Так, эмоциональное возбуждение сопровождается изменением корковой регуляции теплопродукции, углеводного обмена и др. Многие нарушения обмена веществ, терморегуляции, полового и физического развития обусловлены поражением промежуточного мозга. Особенно велика роль гипоталамуса, который с помощью рилизинг-факторов (либеринов и статинов) через гипофиз или парагипофизарные проводниковые пути оказывает влияние на метаболизм. Нарушения вегетативной нервной системы также вызывают изменения в обмене веществ. С поражением симпатических узлов, спинного и промежуточного мозга связано возникновение болезни Барракера-Симмонса. Таким образом, между внутриклеточными механизмами саморегуляции, связанными с генетической информацией и нервно-гормональной регуляцией обмена веществ, имеется тесная связь и нарушение любого из них сопровождается развитием патологии. Нарушения энергетического обмена Нарушения обмена энергии лежат в основе большинства функциональных и органических нарушений органов и тканей. Они могут возникать на всех этапах энергетических превращений вследствие отсутствия или недостатка субстрата, изменения количества или активности ферментов, в связи с генетическими дефектами, действием ингибиторов ферментов эндо- и экзогенного происхождения, недостаточным поступлением в организм незаменимых аминокислот, жирных кислот, витаминов, микроэлементов и других веществ, необходимых для осуществления метаболических процессов или в результате повреждения регуляторных систем. Нормальное течение обменных процессов на молекулярном уровне обусловлено динамическим взаимодействием процессов катаболизма и анаболизма. Катаболизм может совершаться внеклеточно с помощью пищеварительных ферментов и внутриклеточно при участии лизосомальных гидролаз. Внутриклеточному распаду подвергаются собственные макромолекулы, имеющие конформационные нарушения, приобретенные в результате случайных ошибок синтеза либо других повреждений, в частности перекисного окисления. Продукты их распада используются клеткой для синтеза других компонентов. Генетическая недостаточность лизосомальных ферментов приводит к возникновению болезней накопления (мукополисахаридозы, сфинголипидозы, гликогенозы). Частным примером внеклеточного распада макромолекул является протеолиз, который обеспечивает повышение функциональной активности ферментов, гормонов, нуклеиновых кислот, первоначально синтезирующихся в форме предшественников с большей молекулярной массой, чем у основной функционально активной молекулы (например, проинсулин – инсулин). Ферментативный процесс такого типа называется ограниченным протеолизом. Характерным примером его является функционирование каскадных систем: системы комплемента, свертывания крови, фибринолиза, кининовой системы. Наиболее эффективным в энергетическом отношении является окисление продуктов обмена в цикле Кребса, менее эффективным – ?-окисление, гликолиз. При нарушении катаболических процессов прежде всего страдает регенерация АТФ, а также поступление необходимых для биосинтетических процессов (анаболизма) субстратов. В свою очередь повреждение анаболических процессов приводит к нарушению воспроизведения функционально важных соединений – ферментов, гормонов, необходимых для осуществления катаболизма. Наиболее выраженные нарушения катаболизма наблюдаются при повреждении системы биологического окисления или механизмов сопряжения дыхания и окислительного фосфорилирования. Примерно на две трети сокращается выработка энергии при блокировании цикла трикарбоновых кислот (ингибирование фермента цитратсинтазы, дефицит пантотеновой кислоты, гипоксия). Ослабление гликолитических процессов, например, при сахарном диабете нарушает использование углеводов, ведет к гипергликемии, переключению энергетики на липиды и белки, угнетению цикла трикарбоновых кислот (дефицит щавелевоуксусной кислоты), усилению распада белков, кетогенезу и т. д. Нарушение гликолитических процессов отрицательно сказывается на возможности организма адаптироваться к гипоксии. Степень сопряженности дыхания и фосфорилирования в клетках является регулируемым процессом, связанным с состоянием митохондрий. В составе митохондриальных мембран имеются контрактильные белки, аналогичные актомиозиновому комплексу, которые обусловливают возможность активного "сокращения" или "набухания" митохондрий (С. А. Нейфах). В патологических условиях при нарушении сократительных свойств, как это бывает в раковых клетках, митохондрии могут длительное время находиться в набухшем состоянии. Это также способствует выходу факторов, стимулирующих гликолиз, усиливающих гликолитический путь обмена в тканях. В некоторых условиях, особенно связанных с необходимостью поддержания постоянной температуры тела, например при действии холода, организм нуждается в срочной мобилизации тепла, которая происходит путем разобщения окислительного фосфорилирования и повышения удельного веса свободного окисления. К разобщающим факторам относятся: паратирин, прогестерон, гормон роста, вазопрессин, некоторые компоненты дыхательной цепи, динитрофенол, урамицидин и др. Особый интерес представляют данные о разобщающем эффекте бактериальной интоксикации – дифтерийного токсина, золотистого стафилококка. Калоригенный эффект тироксина тоже объяснялся разобщением окисления и фосфорилирования. Однако это не подтвердилось, хотя тироксин и вызывает существенные изменения в митохондриях, в том числе и набухание. Предполагается, что повышение теплопродукции при гипертиреозе связано с увеличением массы митохондрий и повышением активности окислительных ферментов. По-видимому, определенный вклад в этот процесс вносит одновременная стимуляция ана- и катаболических процессов, в связи с чем энергия, направляемая на процессы синтеза, бесполезно рассеивается и ресинтез АТФ затрудняется. Окислительное фосфорилирование существенно нарушается при авитаминозах, особенно группы В, поскольку многие из витаминов этой группы входят в состав коферментов цикла трикарбоновых кислот и переноса электронов в дыхательной цепи. При болезни бери-бери, вызванной отсутствием или недостаточностью тиамина, нарушается цикл Кребса и тем самым уменьшается количество субстратного материала для дыхательной цепи. Судороги и психозы, наблюдаемые при этом, являются клиническими симптомами нарушения биологического окисления в мозге. Нарушения в дыхательной цепи, связанные с отсутствием никотинамидных и флавиновых дегидрогеназ, наблюдаются при пеллагре и арибофлавинозе. Биоэнергетические процессы нарушаются при многихвирусных заболеваниях, в частности при вирусном гепатите, когда вирус использует для нужд своего роста ряд жизненно Необходимых веществ (АТФ, АМФ, рибонуклеиновые кислоты, ацетил-СоА и др.). Дефицит рибонуклеиновых кислот приводит к нарушению синтеза белков клетки, в частности клеточных ферментов, а расходование свободных нуклеотидов – к недостаточному образованию НАД и НАДФН. Глубокие нарушения энергетического обмена возникают при диабете. При этом значительно уменьшается выработка макроэргических соединений в связи с нарушением дыхательной цепи, обусловленным ограничением мощности цикла Кребса. Нарушения основного обмена Для того чтобы получить представление о патологических отклонениях в обмене веществ, обычно исходят из величины основного обмена. На величину основного обмена, даже в физиологических условиях, могут оказывать влияние различные факторы. Доказана роль рефлекторных и условно-рефлекторных, а также гормональных влияний на основной обмен. Особенно ярко это проявляется в условиях патологии – при нарушении нейрогормональной регуляции обмена. Так, у психически больных в стадии прогрессивного паралича и старческого слабоумия находили умеренное снижение основного обмена. Более резкие нарушения его наблюдались при поражении вегетативных диэнцефальных центров (диэнцефалический синдром Пэйджа, опухоли, кровоизлияние в мозг). Особую роль в регуляции основного обмена играет гормон щитовидной железы – тироксин, который является одним из основных регуляторов проницаемости митохондрий, оказывающий влияние на процесс окисления и фосфорилирования и, следовательно, на интенсивность энергетических процессов. Повышение основного обмена на 20% и более является важным диагностическим признаком тиреотоксикоза, а снижение его свидетельствует о гипофункции щитовидной железы. Определенное влияние на основной обмен оказывают гормоны гипофиза. Соматотропин, например, стимулирует свободное окисление и тем самым повышает теплообразование, чем объясняется усиление энергетических процессов при опухолях гипофиза (например, при эозинофильной аденоме). В то же время гипофункция гипофиза, сопровождаясь уменьшением продукции тиротропина и кортикотропина, приводит к снижению теплопродукции и основного обмена. Выраженным стимулирующим действием на основной обмен обладает адреналин, причем этот эффект особенно проявляется в условиях холода. Инсулин обладает противоположным влиянием, он ослабляет мышечную дрожь и теплопродукцию, увеличивая сопряжение окисления и фосфорилирования. У людей, страдающих аддисоновой болезнью (двустороннее повреждение надпочечных желез, обычно туберкулезного происхождения), энергетические процессы угнетаются. Половые гормоны – тестостерон и прогестеронактивизируют свободное окисление и способствуют освобождению энергии. При гипофункции половых желез (кастрация, недоразвитие, климакс) интенсивность энергетических процессов снижается, что сопровождается снижением основного обмена и нередко ожирением. Повышение основного обмена может наблюдаться при усилении сердечной деятельности и дыхания. В начальной стадии развития недостаточности сердца повышение основного обмена составляет 30 – 50%. В патогенезе этого явления участвует гипоксия, которая вызывает компенсаторное усиление работы органов дыхания и кровообращения. Образующаяся при этом молочная кислота частично окисляется с дополнительными затратами кислорода. Гиперкапния тоже возбуждает дыхание и усиливает сердечную деятельность с увеличением основного обмена. Повышение основного обмена при лихорадке объясняется разобщением окисления и фосфорилирования. При голодании основной обмен снижается в связи с переходом организма на экономное расходование энергии. Нарушения углеводного обмена Патология углеводного обмена может быть представлена совокупностью нарушений катаболических и анаболических превращений углеводов. Нарушения катаболизма углеводов могут возникать в результате нарушения переваривания и всасывания углеводов в кишках, гликонеогенеза и гликогенолиза в печени и дальнейшего превращения глюкозы в пировиноградную кислоту, катализируемого ферментами гликолиза. Нарушение ферментативного расщепления полисахаридов в кишках встречается сравнительно редко, поскольку амилаза вырабатывается слюнными, кишечными и поджелудочной железами. При ахилии действие амилазы слюны продолжается и в желудке. В некоторых случаях, особенно при нарушении гормональной регуляции, воспалении слизистой оболочки, отравлениях ядами (монойодацетатом, флоридзином) могут нарушаться процессы всасывания моносахаридов в кишках. Большинство моносахаридов в клетках кишок фосфорилируется гексокиназой. Нарушение процессов фосфорилирования неблагоприятно сказывается на всасывании углеводов. Нарушения анаболизма углеводов проявляются нарушениями синтеза и депонирования гликогена в печени. При миастении, гипоксии отмечается нарушение гликогенеза. При охлаждении, перегревании, боли, судорогах, эмоциях может усиливаться гликогенолиз, при сахарном диабете – гликонеогенез. Существенные нарушения в углеводном обмене возникают при авитаминозах, особенно группы В, поскольку эти витамины являются коферментами многих важных ферментов. Нарушение нервно-гормональной регуляции является наиболее частой причиной патологии углеводного обмена. Объектами регуляции являются три основных процесса углеводного обмена: отложение углеводов в печени и мышцах в форме гликогена и переход их в жиры, т. е. депонирование в качестве источника энергии; гликогенолиз, гликонеогенез и поступление в кровь глюкозы; расщепление глюкозы с освобождением энергии. Эти процессы тесно связаны между собой. Нарушение этой координации проявляется в видегипер- или гипогликемии и гликозурии. Последствия нарушения нервной регуляции углеводного обмена впервые были продемонстрированы Клодом Бернаром (1855), который показал, что укол в дно IV желудочка приводит к гипергликемии. К гипергликемии может приводить раздражение серого бугра гипоталамуса, чечевичного ядра и полосатого тела базальных ядер большого мозга. Кеннон наблюдал, что психическое перенапряжение, эмоции могут повышать содержание глюкозы в крови. Гипергликемия возникает также при болевых ощущениях, во время приступов эпилепсии и т. д. Имеется и второй путь центрального влияния нервной системы на углеводный обмен, которое распространяется к панкреатическим островкам по парасимпатическим волокнам. Нарушение гормональной регуляции углеводного обмена может возникать не только при нарушении центральных механизмов регуляции деятельности соответствующих эндокринных желез, но и при патологии самих желез или же при нарушении периферических механизмов действия гормонов. Ведущим фактором в нарушении гормональной регуляции обмена углеводов является изменение соотношения между активностью инсулина и контринсулярных гормонов. На рис. 14.1 показано влияние инсулина и его антагонистов на процессы, обусловливающие утилизацию глюкозы в тканях и определяющие, в известной мере, уровень гликемии.  Дефицит инсулина и преобладание контринсулярных гормонов сопровождается гипергликемией. Ее происхождение объясняется снижением аллостерического эффекта инсулина, что приводит к снижению клеточной проницаемости для глюкозы, замедлению скорости гексокиназной реакции и образования гексозо-6-фосфата, а следовательно, и дальнейшего метаболизма глюкозы, усилением процессов гликонеогенеза. Гипергликемия наблюдается также при избыточном содержании глюкагона, адреналина, тиреоидина, гликокортикоидов, самототропина и кортикотропина в крови.Глюкагон усиливает гликогенолиз в печени. Он оказывает гликонеогенетическое, липолитическое и инсулинстимули-рующее действие, принимает участие в патогенезе сахарного диабета. У больных акромегалией с пониженной толерантностью к углеводам наблюдается повышенный уровень глюкагона в крови. Гипергликемия также является результатом гликогенолиза в печени. Обычно адреналиновая гипергликемия не длительна, однако при опухолях мозгового вещества надпочечников (феохромоцитоме) она более постоянна. К группе контринсулярных гормонов относятся такжегликокортикоиды, которые, индуцируя синтез матричной РНК, ответственной за Образование белков – ферментов гликонеогенеза, способствуют повышению уровня гликемии. В противоположность инсулину гидрокортизон понижает проницаемость клеточных мембран и замедляет скорость гексокиназной реакции. Гликокортикоиды принимают участие в механизме возникновения гипергликемии при сахарном диабете и болезни Иценко-Кушинга. Кортикотропин действует аналогично гликокортикоидам, так как, стимулируя их выделение, усиливает гликонеогенез и тормозит активность.гексокиназы. Повышенная продукция гормона аденогипофиза -соматотропина (гормон роста), например при акромегалии, сопровождается пониженной толерантностью к углеводам и гипергликемией. Существует представление о том, что соматотропин вызывает гиперплазию ?-клеток панкреатических островков и увеличивает секрецию глюкагона. Наряду с гликокортикоидами соматотропин снижает активность гексокиназы и, следовательно, потребление глюкозы тканями, т. е. является также контринсулярным гормоном. Кроме того, соматотропин стимулирует активность инсулиназы печени. Введение его животным повышает функцию ?-клеток панкреатических островков, что может привести к истощению их и возникновению метагипофизарного диабета. Гормоны щитовидной железы также участвуют в регуляции углеводного обмена. Известно, что гиперфункция щитовидной железы характеризуется понижением устойчивости организма к углеводам. Тироксин стимулирует всасывание глюкозы в кишках, а также усиливает активность фосфорилазы печени. Гипергликемия может быть алиментарной. Если активность инсулина преобладает над активностью контринсулярных гормонов, то в углеводном обмене усиливаются анаболические процессы и устанавливается гипогликемия. Наиболее выраженная гипогликемия бывает при инсулиноме, избыточном введении инсулина извне. Гипогликемия наблюдается при опухолях гипоталамуса, гипофункции гипофиза, аддисоновой болезни. Она возможна при углеводном голодании, тяжелой мышечной работе (марафонский бег), поражении клеток печени, гликогенозах. При снижении уровня глюкозы в крови менее 2,5 ммоль/л возможно развитие гипогликемической комы. Кома – это патологическое торможение центральной нервной системы, характеризующееся потерей сознания, отсутствием рефлексов и расстройством регуляции жизненно важных функций организма. В патогенезе гипогликемической комы основное значение имеет снижение утилизации глюкозы клетками головного мозга, для деятельности которых глюкоза является основным энергетическим источником. Коме обычно предшествует появление голода, в связи с возбуждением вентролатеральных ядер гипоталамуса, тахикардия (гиперпродукция адреналина), усиление потоотделения, слабость, раздражительность, а затем могут развиться судороги. Соотношения между действием различных гормонов на регуляцию глюкозы в крови представлены на рис. 14.2. 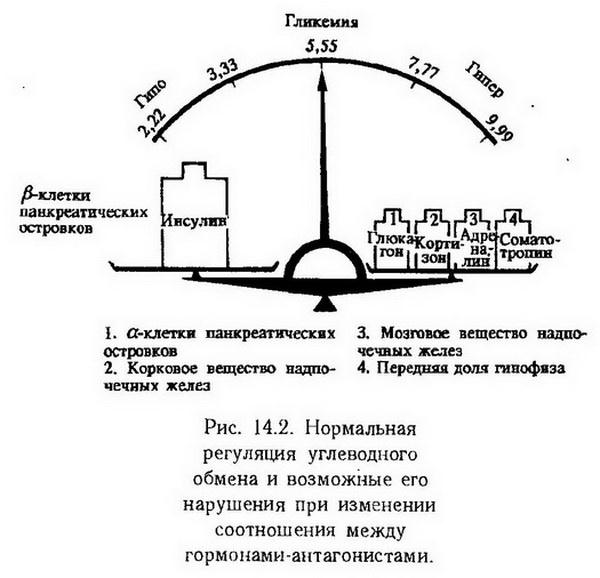 О состоянии регуляции углеводного обмена, о способности организма усваивать определенное количество углеводов судят по толерантности к углеводам, которую определяют с помощью глюкозной нагрузки. Скорость снижения уровня глюкозы во многом зависит от функции панкреатических островков. На рис. 14.3 показаны кривые, отражающие нагрузки глюкозой в норме и при пониженной толерантности к углеводам, которые могут быть при сахарном диабете, акромегалии, базедовой болезни, синдроме Иценко-Кушинга. В этих случаях максимум подъема кривой бывает выше 8,5 ммоль/л, а нормализация ее не завершается к концу 3 – 4 ч. Повышенная толерантность к углеводам наблюдается при гиперфункции панкреатических островков, микседеме. Гликемическая кривая в этих случаях характеризуется незначительным максимальным подъемом и резко выраженной гипогликемией ко второму часу наблюдения. 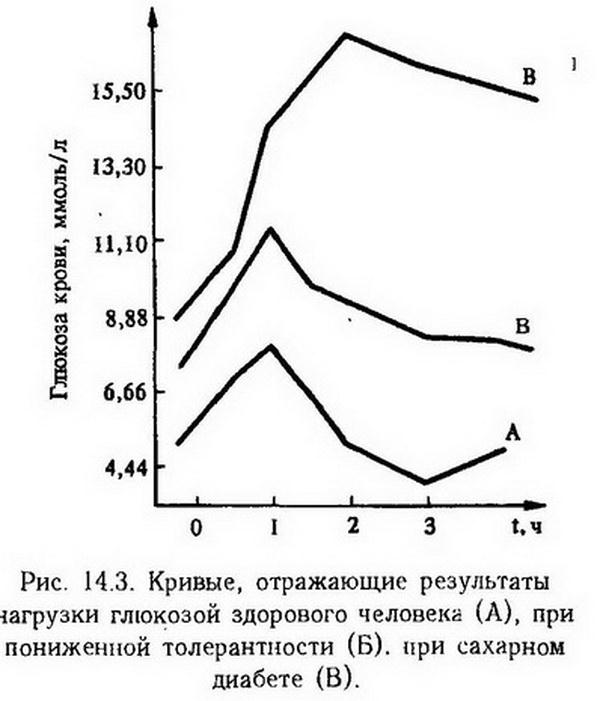 Толерантность к углеводам определяет то максимальное количество глюкозы, которое организм может усвоить без появления гликозурии. У человека это составляет 160 – 180 г глюкозы, принятой натощак. При пониженной толерантности к углеводам гликозурия развивается и от меньшего количества потребленной глюкозы. В общем гликозурия появляется тогда, когда уровень глюкозы в крови превышает почечный порог- 8,8 ммоль/л. При большой концентрации глюкозы в крови ферментативные системы, ответственные за процесс реабсорбции глюкозы в почечных канальцах (гексокиназа, фосфатаза), не обеспечивают фосфорилирование всей глюкозы и часть ее выделяется с мочой. В некоторых случаях гликозурия появляется и без гипергликемии. Это бывает связано с нарушением процесса фосфорилирования глюкозы в почках, например, при введении флоридзина (гликозида из коры фруктовых деревьев), который ингибирует фосфорилирование. При нарушении ферментативных процессов в почках, лежащих в основе реабсорбции глюкозы, развивается почечный диабет. Сахарный диабет Сахарный диабет – это состояние хронической гипергликемии, обусловленной недостаточностью инсулина или избыточностью факторов, противодействующих его активности.Проявления диабета включают нарушения обмена веществ, особенно углеводного, кетоацидоз, прогрессирующее поражение капилляров почек, сетчатки, поражения периферических нервов и выраженный атеросклероз (ВОЗ, 1980). Сахарный диабет встречается у 1 – 4% населения. Среди пожилых – в 2 – 30%. В разных регионах распространенность заболевания неодинакова – от нуля у жителей высокогорных районов Новой Гвинеи до 25% среди индейцев пима (США). Основные проявления диабета – гипергликемия, достигающая иногда 25 ммоль/л, гликозурия с содержанием глюкозы в моче до 555 – 666 ммоль/сут (100 – 120 г/сут), полиурия (до 10 – 12 л мочи в сутки), полифагия и полидипсия. Экспериментальный сахарный диабет. Основные сведения об этиологии и патогенезе сахарного диабета стали известны благодаря опытам на животных. Первая экспериментальная модель его была получена Мерингом и Минковским (1889) путем удаления у собак всей или большей части (9/10) поджелудочной железы. Эта форма экспериментального диабета характеризовалась всеми признаками, наблюдающимися у человека, но протекала более тяжело; всегда осложнялась высокой кетонемией, жировой инфильтрацией печени, развитием диабетической комы. В результате удаления всей поджелудочной железы организм страдал не только от инсулиновой недостаточности, но и от дефицита пищеварительных ферментов. Широкое распространение получила модель аллоксанового диабета, возникающего при введении животным аллоксана. Это вещество избирательно повреждает |3-клетки панкреатических островков, в связи с чем развивается инсулиновая недостаточность различной тяжести. Другим химическим веществом, вызывающим сахарный диабет, является дитизон, связывающий цинк, участвующий в депонировании и секреции инсулина. Повреждает панкреатические островки антибиотик стрептозотоцин. Сахарный диабет у животных может быть получен с помощью антител к инсулину. Такой диабет возникает как при активной, так и пассивной иммунизации. Экспериментальный диабет развивается также при введении контринсулярных гормонов. Так, после длительного введения гормонов передней доли гипофиза (соматотропина, кортикотропина), как отмечено выше, может развиваться гипофизарный диабет. Введением гликокортикоидов можно добиться развития стероидного диабета. Спонтанный диабет встречается у китайских хомячков, мышей линии ККА, OB, AB, мышей "колючих", новозеландских. Все формы экспериментального диабета отчетливо показывают, что в основе развития этого заболевания лежит абсолютная или относительная инсулиновая недостаточность. Этиология. Причиной сахарного диабета является инсулиновая недостаточность. По механизму возникновения инсулиновая недостаточность может быть панкреатической, т. е. связанной с нарушением биосинтеза и выделения инсулина, или внепанкреатической (относительной) при нормальном выделении инсулина панкреатическими островками. Инсулиновая недостаточность может быть связана с генетическими либо приобретенными факторами. Панкреатическая инсулиновая недостаточность возникает в результате нарушений на любой стадии образования и секреции инсулина (рис. 14.4). Причиной инсулиновой недостаточности может быть повреждение глюкорецепторной системы, когда прекращается выброс инсулина в кровь в ответ на раздражение глюкозой мембраны ?-клеток. Инсулиновая недостаточность может быть результатом нарушения механизма поступления кальция в клетку, что затрудняет передачу информации с рецептора в клетку; повреждения аденилатциклазной системы (аллоксан угнетает циклический 3,5-АМФ), гликолиза. К таким же последствиям приводят врожденные и приобретенные поломки в соответствующей части генетического аппарата; дефицит необходимых для синтеза инсулина аминокислот, особенно лейцина и аргинина; нарушения перехода проинсулина в инсулин и секреции инсулина ?-гранулами ?-клеток. Нарушение целости панкреатических островков при различных деструктивных процессах, обусловленных опухолями, кистами, травмами, цирротическими и воспалительными процессами в поджелудочной железе, склеротическом повреждении сосудов также вызывает недостаточность инсулина. Подобную же роль может играть инфекционное поражение островков при скарлатине, коклюше, эпидемическом паротите, гриппе, ангине, роже, сифилисе, туберкулезе. Повреждение островков может явиться результатом иммунной реакции на инсулин либо ?-клетки. Цитотоксичностью по отношению к р-клеткам обладают родентициды, стрептозотоцин, нитрозамины, некоторые лекарственные препараты, диуретики, пероральные контрацептивы, ?-адренэргетики, кортикотропин. Причиной инсулиновой недостаточности может быть употребление пищевых веществ, содержащих цианиды (корни маниока, сорго, ямс). Обычно цианиды обезвреживаются при участии серосодержащих аминокислот, но при белковой недостаточности создаются условия для их накопления в организме. Таково происхождение сахарного диабета при квашиоркоре и других формах недостаточности питания у жителей стран тропического пояса. 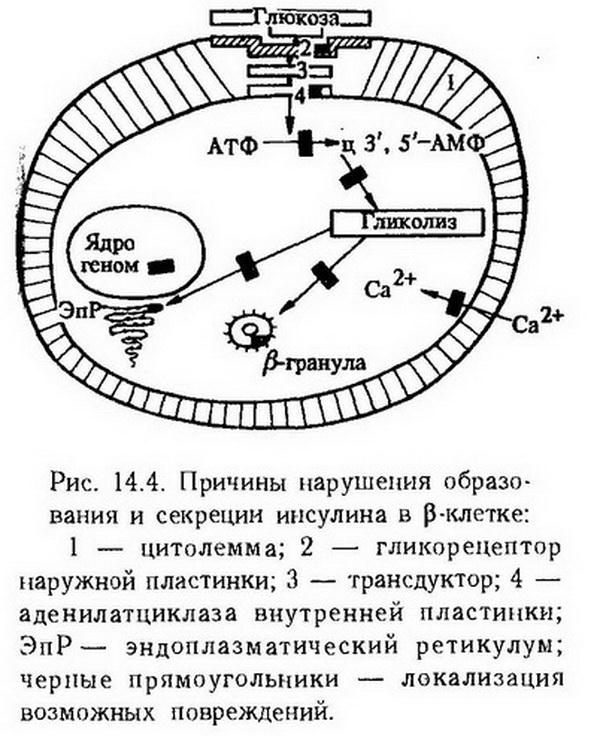 Внепанкреатическая или относительная инсулиновая недостаточность возникает в тех случаях, когда появляются факторы, угнетающие действие инсулина или ускоряющие его катаболизм. К ним можно отнести повышенную продукцию контринсулярных гормонов. При сахарном диабете секреция глюкагона продолжается, несмотря на высокую гликемию, что можно объяснить дефектами глюкозорецепторов ?-клеток. К инактивации инсулина могут привести повышенная активность инсулиназы, хронические воспалительные процессы, сопровождающиеся поступлением в кровь протеолитических ферментов, избыток в крови неэстерифицированных жирных кислот, антитела. Антитела образуются к экзогенному и эндогенному инсулину в ответ на структурные изменения инсулина, обусловленные генетическими нарушениями синтеза белков, соматической мутацией бета-клеток. Нарушение структуры инсулина может быть причиной, снижающей его физиологическую активность. К внепанкреатической инсулиновой недостаточности могут привести отсутствие ферментов, освобождающих инсулин от связи с сывороточными белками, антагонисты инсулина, например синальбумин. Предполагают, что синальбумин является В-цепью инсулина, он взаимодействует с рецепторами инсулина на клеточной мембране, приводя к относительной недостаточности гормона. Наконец, в ряде случаев заболевание обусловлено состоянием инсулиновых рецепторов. При сахарном диабете может снижаться число рецепторов или их сродство к инсулину, возможно образование антител, блокирующих связывание инсулина с рецептором. В некоторых случаях повреждение рецепторов может быть первичным генетическим нарушением. Снижение физической активности, избыточное питание и последующее ожирение также могут вызвать резистентность к инсулину. В одних случаях при ожирении снижается количество рецепторов инсулина на клетках-мишенях, в других возникают пострецепторные изменения, например, вследствие снижения транспорта глюкозы или затруднения ее внутриклеточного метаболизма. На рис. 14.5 представлены основные возможные причины панкреатической и внепанкреатической недостаточности инсулина. 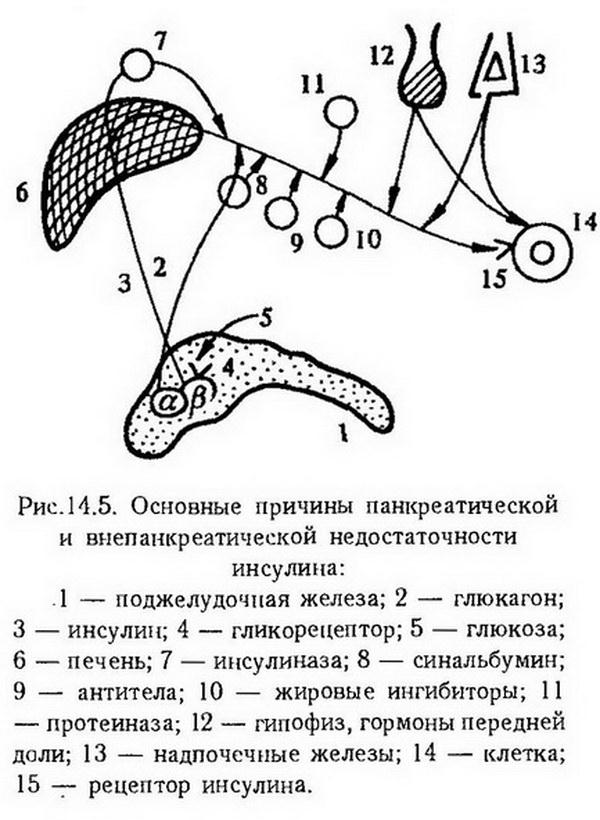 Развитие диабета может быть связано с наследственной неполноценностью панкреатических островков, которая проявляется при воздействии таких провоцирующих факторов, как инфекция, интоксикация, физические и психические травмы, избыточное потребление с пищей жиров и углеводов. Наследоваться могут генетические дефекты, касающиеся строения мембран ?-клеток, их энергетики, различных этапов синтеза инсулина с образованием аномального процесса выделения инсулина, содержания инсулиновых антагонистов, структуры рецепторов инсулина и др. Наследственная форма диабета встречается у мышей, причем в одних случаях он наследуется по аутосомно-рецессивному типу, в других – по доминантному. В гомозиготе ген детален. Роль генетических факторов в этиологии диабета подтверждается наличием семейного диабета, когда заболеваемость сахарным диабетом регистрируется у нескольких членов одной семьи, иногда в 3 – 4 поколениях; высокой конкордантностью у монозиготных близнецов (в 4 раза выше, чем у дизиготных). По-видимому, имеется несколько мутантных генов, определяющих развитие сахарного диабета. Наследование может осуществляться аутосомно-рецессивно, доминантно и полигенно. Поскольку риск заболеваемости для женщин в 2 раза выше, чем для мужчин, высказывается предположение о связи патологического гена с 10-й хромосомой; с антигенами системы АВО (чаще заболевают люди, имеющие II группу крови). Наиболее убедительные данные получены о связи инсулинзависимого сахарного диабета (ИЗСД) с определенными лейкоцитарными антигенами. Классификация. Сахарный диабет в зависимости от причин и степени инсулиновой недостаточности может быть первичным и вторичным (симптоматическим). В первичном сахарном диабете, по предложению экспертов ВОЗ (1980, 1987), выделен инсулинзависимый (диабет I типа) и инсулиннезависимый сахарный диабет (диабет II типа). Другие типы диабета являются вторичными и связаны с определенными заболеваниями, например, акромегалией, болезнью Иценко-Кушинга, болезнями поджелудочной железы, действием лекарственных, химических средств, генетическими синдромами и др. Отдельные классы представляет диабет, связанный с недостаточностью питания, и диабет беременных. Наибольшее распространение имеет первичный диабет. В его происхождении наряду с генетическими факторами определенную роль играют иммунные механизмы и внешнесредовые влияния, однако соотношение между ними при диабете I и II типа различно. Инсулинзависимый сахарный диабет (ИЗСД) характеризуется инсулинопенией, т. е. абсолютной инсулиновой недостаточностью, существенными метаболическими нарушениями с тенденцией к кетоацидозу. Развивается эта форма диабета в юношеском возрасте, генетическая предрасположенность сочетается с определенными антигенами системы HLA. Чаще всего среди европейской популяции встречается HLA-B8-DR3 и B15-DR4. Имеются также факты, указывающие на аутоиммунное происхождение диабета этого типа: сочетание с другими аутоиммунными заболеваниями (инсулинит, тиреоидит), наличие антител к инсулину, ?-клеткам. Нередко начало заболевания связано с предшествующей вирусной инфекцией (корь, краснуха, гепатит, эпидемический паротит). Взаимосвязь генетических и иммунных механизмов объясняется расположением генов системы HLA вблизи локуса, ответственного за иммунный ответ организма, на 6-й хромосоме. Очевидно, наличие определенных лейкоцитарных антигенов свидетельствует об особенностях иммунного статуса организма. Внешним фактором, способствующим реализации наследственной предрасположенности к сахарному диабету, могут быть вирусы, обладающие повышенной бета-тропностью у лиц, носителей антигенов системы HLA. Вирусы разрушают ?-клетки либо, повреждая их, дают начало аутоиммунному процессу. Инсулиннезависимый сахарный диабет (ИНСД) характеризуется минимальными метаболическими нарушениями. В его основе лежит относительная инсулиновая недостаточность, провоцируемая перееданием, ожирением, уменьшением числа рецепторов к инсулину. Антитела к (3-клеткам и инсулину отсутствуют. Диабет проявляется после 40 лет и, несмотря на отсутствие генетических маркеров в виде лейкоцитарных антигенов, имеет более выраженную наследственную обусловленность. Конкордантность у монозиготных близнецов достигает 90 – 100%, в то время как при диабете I типа – только до 50%. Патогенез. Инсулиновая недостаточность при сахарном диабете сопровождается нарушением всех видов обмена, прежде всего углеводного, проявлением чего являются гипергликемия и гликозурия. Одним из основных патогенетических механизмов нарушений углеводного обмена при сахарном диабете является замедление скорости гексокиназной реакции, обусловленное снижением проницаемости клеточных мембран и, следовательно, транспорта глюкозы в клетки, а также, возможно, понижением активности гексокиназы в клетках, в частности печени, свободно проницаемых для глюкозы. Это в свою очередь приводит к замедлению образования глюкозо-6-фосфата (Г-6-Ф), а затем и использования этого первого метаболита обмена глюкозы на всех путях превращения его в клетке – синтез гликогена, пентозофосфатный цикл и гликолиз. В печени дефицит Г-6-Ф компенсируется образованием его в процессе гликонеогенеза. Повышение активности фосфорилазы и глюкозо-6-фосфатазы печени способствует усилению глюкозообразования и понижению образования гликогена в ней. При сахарном диабете отмечается активация гликонеогенеза, которая объясняется относительным преобладанием гликокортикоидов, индуцирующих синтез необходимых для этого ферментов. Таким образом, замедление скорости гексокиназной реакции, усиление гликонеогенеза и повышение активности глюкозо-6-фосфатазы являются главными причинами диабетической гипергликемии. С. Г. Генес считает, что гипергликемия при сахарном диабете имеет компенсаторный характер, поскольку при высоком уровне глюкозы в крови потребление ее тканями улучшается. В то же время гипергликемия имеет и отрицательное значение, являясь одним из патогенетических факторов диабетических ангиопатий. Ангиопатии возникают у большинства больных сахарным диабетом с длительным течением и неполной компенсацией инсулиновой недостаточности. Они проявляются в виде склероза, облитерации и других поражений кровеносных сосудов. Ведущими факторами в генезе этих осложнений являются генетическая предрасположенность, гиперпродукция контринсулярных гормонов и метаболические сдвиги, в частности гипергликемия и гиперхолестеринемия. Гипергликемия сопровождается повышением концентрации глико- и мукопротеидов, которые легко выпадают в соединительной ткани, способствуя образованию гиалина и поражению сосудистой стенки. Предполагают, что в этом процессе принимают участие аутоиммунные механизмы. Гипергликемия и нарушение процессов фосфорилирования и дефосфорилирования глюкозы в канальцах нефрона приводят к гликозурии. Повышение осмотического давления мочи способствует полиурии, которая в свою очередь вызывает обезвоживание организма и, как следствие его, усиленную жажду (полидипсия). Нарушение жирового обмена при инсулиновой недостаточности характеризуется снижением образования жира из углеводов и ресинтеза триглицеридов из жирных кислот в жировой ткани. Усиление липолиза и выход жирных кислот из жировой ткани связаны с относительным усилением липолитического эффекта соматотропина. Усиливается также окисление жирных кислот в тканях. Жирные кислоты в увеличенном количестве поступают в печень, которая сохраняет способность к синтезу триглицеридов, что и создает предпосылку к ожирению печени. Однако ожирение может не наступить, если в поджелудочной железе не нарушено образование липокаина (см. ниже – "Нарушения жирового обмена"). При сахарном диабете происходит усиленное образование в печени и накопление в организме кетоновых тел. Объясняется это явление тем, что образовавшийся в большом количестве при расщеплении жирных кислот ацетил-СоА не может полностью превратиться в цитрат и сгореть в цикле Кребса, поскольку метаболическая мощность последнего при диабете ограничена и включение в него активной уксусной кислоты нарушено. Кроме того, при этом замедлен ресинтез жирных кислот из ацетил-СоА в печени, жировой и других тканях в результате дефицита НАДФН2, возникшем в связи со снижением скорости пентозофосфатного цикла. Снижена также активность ферментов, синтезирующих жирные кислоты через малонил-СоА. Поэтому неиспользуемый в синтезе жирных кислот и невовлекаемый в цикл Кребса ацетил-СоА является источником усиленного кетогенеза и синтеза холестерина. Имеются данные и об участии в этом процессе глюкагона, который стимулирует фермент карнитинацетилтрансферазу, ускоряющую окисление жирных кислот с образованием кетоновых тел. Нарушение белкового обмена при сахарном диабете проявляется не только угнетением анаболических процессов, но и усилением катаболизма белков с использованием дезаминированных аминокислот для образования углеводов (гликонеогенез). Замедление синтеза белка и ускорение гликонеогенеза способствуют развитию отрицательного азотистого баланса. В связи с нарушением белкового обмена угнетаются пластические процессы, заживление ран, выработка антител. Нарушение гормональной регуляции, сопровождающей сахарный диабет, может отражаться на течении беременности, приводит к патологии плаценты, самопроизвольным абортам. В связи с нарушением проницаемости плаценты для гормонов, развитием гипоксии возникают эмбриопатии, приводящие к выраженным порокам развития. Грозным осложнением сахарного диабета являетсядиабетическая кома – результат интоксикации организма кетоновыми телами. Кому, основными проявлениями которой являются потеря сознания, патологическое дыхание типа Куссмауля, снижение артериального давления, называют гиперкетонемической или гипергликемической. В последние годы у больных сахарным диабетом наблюдается кома, развивающаяся при отсутствии кетоновых телец, но при очень высокой гипергликемии (до 50 ммоль/л и более) и значительном повышении в крови содержания натрия, мочевины и хлора. Все это ведет к гиперосмии и дегидратации. Кома получила название гиперосмолярной. Отсутствие кетоацидоза объясняется сохранением остаточной секреции инсулина, достаточной для нормализации углеводного и водно-электролитного обмена. Наследственные нарушения углеводного обмена Как уже было сказано выше, в возникновении нарушений углеводного обмена (сахарного диабета) большое значение придают наследственности. Наследственные нарушения углеводного обмена могут быть обусловлены недостаточностью специфических ферментов или транспортной системы мембраны, необходимых для обмена определенного сахара: клинические проявления при этом варьируют от доброкачественной пентозурии у практически здорового ребенка до галактоземии, когда больному грозит гибель от истощения и печеночной недостаточности или тяжелой диареи и дегидратации при синдроме нарушенного всасывания глюкозы и галактозы. В основе синдрома нарушения всасывания углеводов лежит недостаточность специфической дисахаридазы щеткообразного эпителия либо системы транспорта моносахаридов. В обоих случаях сахар накапливается в просвете кишечника, повышая осмолярность кишечного сока и тем дополнительно насасывая в просвет кишечника воду. Дети страдают от болей и вздутия живота, поноса, они отстают в росте и развитии. Среди других наследственных дефектов углеводного обмена следует назватьгалактоземию – рецессивно наследуемое заболевание, проявляющееся в неспособности к обмену галактозы, входящей в состав лактозы молока. Наследственным биохимическим дефектом при этом является секреция в печени и эритроцитах галактозо-1-фосфатуридил-трансферазы с нарушенной активностью. Галактоземия Галактоземия сопровождается галактозурией. Обмен галактозы задерживается на уровне галактозо-1-фосфата, который, как и галактоза, накапливается в крови, селезенке, печени, хрусталике. Развиваются катаракта, цирроз печени. У детей отмечаются задержка развития, исхудание, умственная отсталость. Исключение галактозы из пищи сохраняет ребенку жизнь. Наследственно пониженная активность любого из ферментов распада или синтеза гликогена приводит к развитию гликогенозов, которые относятся к болезням накопления. Самым распространенным гликогенозом является болезнь Гирке, возникающая в результате дефицита глюкозо-6-фосфатазы. Заболевание у детей проявляется избыточным отложением гликогена в печени и почках, гипогликемией. Отмечается отсталость физического и психического развития. При болезни Герса имеют место низкая активность фосфорилазы и отложение гликогена в печени и лейкоцитах. Диффузный гликогеноз (болезнь Форбса) характеризуется появлением гликогена с короткими многочисленными внешними ветвями. Это заболевание связывают с пониженной активностью амило-1,6-глюкозидазы. У детей, страдающих этой формой гликогеноза, отмечаются отеки, кровоточивость. Гликоген депонируется в печени, мышцах, эритроцитах, лейкоцитах. К рецессивно наследуемым патологическим состояниям относятся также фруктозурия и пентозурия. Существует две формы фруктозурии. Одна из них – наследственная непереносимость фруктозы, связанная с отсутствием фруктозодифосфат-альдолазы в печени, почках, слизистой оболочке кишок, протекает иногда как тяжелое заболевание. Накапливающийся при этом фруктозо-1,6-дифосфат, как полагают, оказывает токсическое действие. После приема фруктозы у больных возникает гипогликемия, происхождение которой не выяснено. Развивается поражение печени, почек. Вторая форма фруктозурии – доброкачественная, протекающая бессимптомно, связана с недостаточностью в печени кетогексокиназы, катализирующей превращение фруктозы в фруктозо-1-фосфат, вследствие чего фруктоза накапливается в крови. Рецессивно наследуемая пентозурия возникает в результате дефицита НАДН-зависимого фермента, восстанавливающего L-ксилулозу до ксилитола. Никаких нарушений в состоянии организма, кроме ежедневного выведения с мочой нескольких граммов L-ксилулозы, не отмечается. Мукополисахаридозы. Для мукополисахаридозов характерно отложение в различных тканях организма полимерных углеводов, называемых глюкозоаминогликанами (ГАГ) или мукополисахаридами. ГАГ входят в состав основного межклеточного вещества. Заболевание обусловлено дефектом специфической лизосомальной гидролазы, принимающей участие в последовательном расщеплении гликозоаминогликанов. Нерасщепленный материал откладывается в лизосомах почти всех клеток. Всем больным свойственны гротескные черты лица, изменение скелета ("множественный дизостоз"), изменения в суставах. Поражаются печень, селезенка, сердце, кровеносные сосуды. Все мукополисахаридозы обусловлены дефектом фермента, принимающего участие в расщеплении дерматансульфата и гепарансульфата. Блок на одном этапе не дает следующему по очереди ферменту осуществить свою реакцию и нерасщепленный материал накапливается в лизосомах. ГАГ входят в перикарион нейронов, аксоны и глию мозга. При патологии не только увеличивается количество ГАГ в мозге, но и меняется соотношение с ганглиозидами. Клиническое проявление – умственная отсталость. Нарушения жирового обмена Патологические изменения в обмене жиров (липидов), в частности триглицеридов и высших жирных кислот, могут возникать в результате нарушения всасывания и выделения жиров; нарушения транспорта жиров в ткани; избыточного накопления жиров в органах, не относящихся к жировой ткани (жировая инфильтрация и жировая дистрофия); нарушения промежуточного жирового обмена; нарушения жирового обмена в жировой ткани (избыточное или недостаточное его образование и отложение). Нарушения всасывания и выделения жиров Одним из основных условий, обеспечивающих нормальное всасывание жира, является его эмульгирование, расщепление на глицерин и жирные кислоты и образование соединений с желчными кислотами (холеинатов). Эмульгирование жира обеспечивается при определенном соотношении желчных, жирных кислот и моноглицеридов. Два последних компонента образуются в результате расщепления жира панкреатической липазой. Следовательно, недостаток липазы, который возникает при заболеваниях поджелудочной железы (панкреатит, острый некроз, склероз), а также дефицит желчных кислот (обтурационная желтуха, билиарный цирроз) сопровождаются нарушением всасывания жира. В этом случае содержание жира в кале резко увеличивается, наблюдаетсястеаторея. Стеаторея может возникать и при использовании антибиотиков (неомицина сульфат и хлортетрациклина гидрохлорид), которые подавляют липолиз. К таким же последствиям приводит понос, вызванный быстрым продвижением жира по кишкам. При избытке в пище кальция и магния нарушается всасывание жирных кислот – образуются нерастворимые в воде соли жирных кислот (мыла), которые выводятся через кишки. Процесс всасывания жиров страдает и при нарушении фосфорилирования. Последнее может измениться при отравлении ядами (монойодуксусная кислота, флоридзин), а также при недостаточности коркового вещества надпочечных желез. Всасывание жира тормозится при поражении эпителия тонких кишок инфекционными и токсическими агентами, при авитаминозах А и В, в течение которых нарушается образование ферментов, участвующих в ресинтезе триглицеридов. На резорбции жира отражается и недостаток холина. В нормальных условиях в организме обычно усваивается около 95% вводимого жира, около 5% жира выводится в основном через кишки и в меньшей степени – сальными и потовыми железами. При приеме жира в больших количествах так же, как и при размножении костного мозга, при травматизации больших участков жировой ткани, липоидном нефрозе, наблюдается липурия Последствием нарушения всасывания жира является качественное голодание (см. раздел XV – "Голодание"). Нарушения транспорта жира и перехода его в ткани Ресинтезированные в кишечной стенке жиры поступают в лимфатическую систему, затем в плечеголовные вены и циркулируют в крови в виде хиломикронов, содержащих до 1% белков и 99% липидов. Первый орган, в котором часть хиломикронов задерживается, – легкие. Они обладают липопектическим свойством, регулирующим поступление жира в артериальную кровь. Если дыхательная функция легких ограничена (пневмоторакс, эмфизема), жиры задерживаются в них. Увеличение дыхательной поверхности легких, ускорение кровотока (у певцов) при недостаточном развитии мезенхиальных элементов легких приводят к большому поступлению липидов в артериальную кровь и отложению их в жировой ткани. Часть хиломикронов в крови расщепляется липопротеидной липазой (фактор просветления), которая локализуется в эндотелии сосудов и выходит в кровь под влиянием гепарина. Образующиеся при этом неэстерифицированные жирные кислоты (НЭЖК) адсорбируются на альбумине и 0-липопротеидах и транспортируются в органы и ткани. В печени часть НЭЖК ресинтезируется в триглицериды, а часть используется как источник энергии. Жиры транспортируются кровью не только от кишок к органам и тканям, но и от жировой ткани к печени и другим органам. На рис. 14.6 представлена схема транспорта и депонирования жиров. 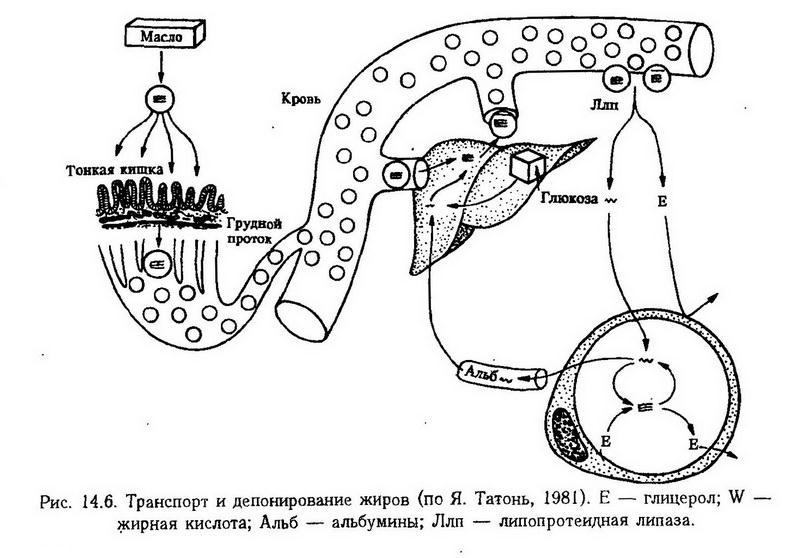 Одним из показателей нарушения жирового обмена является гиперлипемия (свыше 3,5 – 8 г/л). Гиперлипемия может быть алиментарной (пищевой), транспортной (при перемещении жира из депо в печень) и ретенционной (вследствие задержки жира в крови, например, в связи с изменением соотношения белковых фракций крови при постгеморрагической анемии, нефротическом синдроме). Алиментарная гиперлипемия наблюдается через 2 – 3 ч после нагрузки жиром, достигая максимума через 4 – 6 ч. Через 9 ч содержание жира в крови возвращается к норме. Блокада системы мононуклеарных фагоцитов, спленэктомия, нарушение образования гепарина, активирующего липопротеидлипазу, способствуют более высокой и длительной гиперлипемии. Такой же эффект вызывают натрия хлорид, желчные кислоты, которые являются ингибиторами липопротеидлипазы. Те же факторы могут способствовать происхождению ретенционной гиперлипемии. Так, при атеросклерозе гиперлипемия связана с уменьшением в крови содержания гепарина и низкой активностью липопротеидлипазы, при диабете – дефицитом липокаической субстанции и торможением поступления в кровь фермента. Транспортная гиперлипемия возникает при обеднении печени гликогеном (голодание, сахарный диабет), а также при повышенном образовании адреналина, кортикотропина, соматотропина, тироксина и 0-липотропина. Введение глюкозы может тормозить транспорт липидов, поскольку из глюкозы при наличии инсулина синтезируются гликоген и триглицериды. Жировая инфильтрация и дистрофия. Поступающие в ткани жиры подвергаются окислению или депонируются. Если накапливание происходит вне клеток жировой ткани, то говорят о жировой инфильтрации. Сочетание инфильтрации с нарушением структуры протоплазмы жировых клеток определяется как жировая дистрофия. Возможна также жировая декомпозиция, при которой жиры обнаруживаются в клетке в связи с нарушением белково-липидных комплексов. Причиной жировой инфильтрации нередко является снижение активности гидролитических или окислительных ферментов (при отравлении фосфором, мышьяком, хлороформом, при вирусной инфекции, авитаминозах). Чаще всего жировая инфильтрация наблюдается в печени. |