Тест мед ген. Lektsii генетика. Основы клинической генетики
 Скачать 0.6 Mb. Скачать 0.6 Mb.
|

|
Тема 1. Кинико-генеалогический метод. Семиотика наследственных болезней. Клинико-генеалогический метод является основным методом медицинской генетики. Для эффективного применения современных методов диагностики и лечения врожденной и наследственной патологии важнейшим условием является как можно более ранняя постановка точного диагноза. Поэтому владение клинико-генеалогическим методом для современного врача совершенно необходимо. I. Клинико-генеалогический метод (КГМ). КГМ проводится на начальном этапе медико-генетического консультирования. Он основан на детальном сборе анамнестических данных о пробанде и пораженных в семье, а также на генеалогическом и синдромологическом анализе. К общим показаниям для проведения клинико-генеалогического метода относятся: 1) наличие случая наследственного синдрома, а также с наследственной предрасположенностью в семье, либо подозрение по клиническим проявлениям: множественные микро- и макроаномалии у пробанда, особенно в сочетании с отставанием в психо-речевом и моторном развитии; задержка умственного и физического развития; прогрессирующее нарушение функций различных систем организма; - нарушение половой дифференцировки; недоразвитие вторичных половых признаков, задержка или преждевременное половое созревание, сочетающееся с умственной отсталостью или своеобразным психическим дефектом; - нарушение физического развития детей: неправильный рост, деформация костей туловища и конечностей, чрезмерное развитие подкожно-жировой клетчатки, тугоподвижность или гипермобильность в суставах, снижение зрения или слепота, тугоухость и глухота; - нарушение пигментации кожных покровов; - почечно-каменная болезнь у детей; - гемолитическая анемия, желтуха «эндогенного» генеза; - хронический, прогредиентный характер течения заболевания, при котором возникают сложности с установлением диагноза; - первичное невынашивание беременности (1 и > спонтанных абортов, выкидышей, мертворождения); - первичная аменорея, бесплодие у женщин и мужчин; - пренатальная диагностика в случае, если у кого-либо из родителей имеет место наличие хромосомного транслокационного синдрома; - подозрение на синдромы с хромосомной нестабильностью; - оценка эффективности лечения, прогноза при лейкозах; - оценка мутагенных воздействий (в случае, если кто-либо из родителей имел место контакт до или в период беременности с химическими, радиактивными и др. мутагенами); - накопление патологического признака в семье. 2) Основные генетические и клинико-параклинические характеристики наследственных болезней обмена веществ: - при большинстве форм имеет место аутосомно-рецессивный тип наследования (чаще встречаются кровнородственные браки); - начало заболевания, как правило, в раннем детском возрасте; характерен "светлый промежуток" до первых клинических проявлений, т.е. когда состояние больного внешне не выявляет признаков болезни; - злокачественный, чаще всего, характер течения с высокой летальностью в детском возрасте; - полиорганность поражения; - неуклонно нарастающие психомоторные расстройства, диффузная гипотония, гипорефлексия (реже - наоборот) при исключении экзогенных причин болезни; - судорожный синдром; - поражение внутренних органов: гепатоспленомегалия, стойкая желтуха; - патология органа зрения (глазное дно: симптом "вишневой косточки", атрофии дисков зрительных нервов, прогрессирующий офтальмопарез, птоз, как правило 2-х сторонний); - непереносимость отдельных пищевых продуктов, лекарственных препаратов, частая рвота, диарея (при отсутствии экзогенных причин), необычный цвет и запах мочи; - тугоподвижность в суставах и др. прогрессирующие деформации опорно-двигательного аппарата (килевидная грудная клетка,"гаргоилизм", вальгусная установка коленных суставов); - ацидоз, аминоацидурия; - аллергический дерматит; - специфические изменения при биохимическом исследовании активности ферментов и накопление аномальных субстратов в тканях и жидких средах организма (на основании тонкослойной хроматографии и др.); - чаще всего - резистентность к терапии; 3) показания для периконцепционной профилактики. При обращении в генетическую консультацию на пробанда (консультируемое лицо) заполняется генетическая карта, в которую вносится следующая информация: 1) паспортные данные - Ф.И.О., дата и место рождения, национальность, место работы и профессиональные вредности, домашний адрес, диагноз при обращении, а также цель обращения (например: уточнение диагноза, определение прогноза для потомства и др.). Далее заполняются данные о ближайших родственниках пробанда (1-я степень родства - родители, сибсы, т.е. родные братья и сестры): Ф.И.О. (для матерей пробанда в т.ч. и девичью), дата и место рождения (уточняется происхождение родителей), национальность, возраст родителей к моменту рождения пробанда, место работы и профессиональные вредности, состояние здоровья, имеется ли родственная связь между родителями пробанда, дополнительные сведения. 2) Далее подробно собирается анамнез жизни пробанда, для чего уточняются следующие моменты: - состояние здоровья родителей пробанда до зачатия (хр. болезни, злоупотребление алкоголем, наркотиками, табакокурение, имелся ли контакт с мутагенами - радиактивные, химические, биологические и др. факторы); - течение беременности: общее число беременностей и их исходы (имелись ли спонтанные и мед. аборты, выкидыши, мертворождения), от какой беременности рожден пробанд, характер течения (имели ли место токсикозы, анемия, угроза прерывания, маточно-плацентарная недостаточность и др.); перенесенные заболевания и на каких сроках (контакт с краснухой, вирусными инфекциями), токсоплазмоз, травмы, хронические инфекции, и характер проводимого лечения; имелся ли контакт с мутагенными и тератогенными факторами и на каких сроках беременности; Rh-совместисть матери и плода; характер шевеления плода; условия жизни матери (стрессовые ситуации, характер питания); доношенная или недоношенная беременность; - течение родов: на каком сроке, продолжительность (скорые, затяжные), предлежание плода, имела ли место родостимуляция (лекарственная, ваккуум-экстракция, наложение акушерских щипцов); имела ли место при рождении асфиксия, ее степень (обвитие пуповиной, цианоз, закричал сразу или нет), состояние по шкале Апгар, вест, рост; на какие сутки приложен к груди, рефлексы новорожденного (сосание активное или нет, крик громкий или вялый), отмечалась ли желтуха новорожденного; на какие сутки выписаны из родильного дома или переведены в отделение неонаталогии; - развитие на первом году жизни: изменение весо-ростовых показателей, двигательной активности (голову держал, сидел, ходил без опоры), психоречевое развитие, характер вскармливания; имелись ли частые срыгивания, запоры, беспокойство, аллергозы; характер прорезывания зубов; - дальнейшее психомоторное развитие: бегал, прыгал, первые слова, фразы в зависимости от периода - 1-й пубертатный, 2-й пубертатный, юношеский и т.д.; - перенесенные заболевания, в каком возрасте, подробности диагности- ки, эффективности лечения, последствий. Детальный сбор анамнеза жизни пробанда очень важен в плане дифференциальной диагностики наследственных и ненаследственных патологических состояний. 3) Анамнез болезни: возраст и характер начала (острое, постепенное), имелся ли "светлый" промежуток до дебюта; характер течения (стационарное, с положительной динамикой, медленно прогрессирующее, злокачественное); проявления заболевания (подробно), результаты проводимого обследования и лечения. 4) Составление родословной производится путем опроса пробанда и его родственников, а также применяется метод анкетирования, осмотр фотографий родственников, проводится изучение выписок из историй болезни и амбулаторных карт. Чем больше членов семьи будет опрошено, тем полнее будет генеалогическая информация. Существует графическая и прописная методика составления родословных. Родословную начинают составлять с середины нижней трети чистого листа, с пробанда (рис.1). рис.1   1 2 3 4 1 2 3 4 I            1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8 9 10I     I I    1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8 9 10I  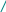 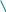 II II  Далее уточняются генеалогические данные о сибсах пробанда (т.е. о родных сестрах и братьях), фиксируются имевшие место спонтанные аборты, выкидыши, мертворождения. Затем, в левой половине родословной, собирается генеалогия по материнской линии пробанда, о ее сибсах, потомстве сибсов, родителях матери пробанда. Таким же образом собирается генеалогическая информация по линии отца пробанда. Если патологический признак прослеживается, то желательно как можно подробнее зафиксировать генеалогические данные, если не прослеживается, то достаточно трех поколений. При составлении родословной принята следующая символика (рис.2): рис.2  - мужчина, - женщина, - пораженнные; - мужчина, - женщина, - пораженнные;     - пробанд; - пол неустановлен; - пробанд; - пол неустановлен;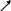 ! , !-лично осмотрены;       - выкидыш, - аборт, - мертворожденные - выкидыш, - аборт, - мертворожденные   - брак, - кровнородственный брак; - брак, - кровнородственный брак; 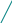   - внебрачная связь, - распавшийся брак; - внебрачная связь, - распавшийся брак;  - повторный брак, , - бесплодие - повторный брак, , - бесплодие     - сибство, , - гетерозиготные носители при - сибство, , - гетерозиготные носители при а утосомно-рецессивных синдромах; - гетерозиготная носительница при Х-сцепленных рецессивных синдромах; -монозиготные близнецы - дизиготные близнецыПод родословной составляется "легенда", в которую включаются все необходимые дополнения не уместившиеся при изображении. Пример: I3 (3-й член родословной в первом поколении) - Иванов Иван Иванович, 1905 г.р., имелись признаки невральной амиотрофии Шарко-Мари-Тус (НАШМТ): измененная походка, похудание голеней, слабость в кистях рук и т.д. При достаточном объеме генеалогической информации производится анализ родословной, уточняется тип наследования и повторный риск заболевания. Рассмотрим критерии наследования. А. Критерии моногенного наследования. 1) Аутосомно-доминантный (АД) тип наследования (рис.3; примеры: с-м Марфана, хорея Гентингтона, миотония Томсена, прогрессирующая мышечная дистрофия (ПМД) лицелопаточноплечевая и др.): - характерна передача патологического признака по вертикали, т.е. из поколения в поколение; - вероятность передачи мутации от больного родителя потомству составляет 50%; - оба пола поражаются с одинаковой частотой; - больной отец может передать патологию сыновьям; - если оба родителя здоровы (при исключении субклинических проявлений, обусловленных неполной пенетрантностью), но в семье рождается больной ребенок с АД синдромом, то такие случаи обусловлены новыми мутациями в родительской гамете, повторный риск заболевания в семье низкий; - мутации в гомозиготном состоянии (встречаются крайне редко), как правило летальны.  рис.3 рис.3 I 1 2 3 4 5 I 1 2 3 4 5      II  1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8 9 10   III 1 2 3 4 5 6 7 8 9 10 2) Аутосомно-рецессивный (АР) тип наследования (рис.4; примеры: атаксия Фридрейха, спинальная мышечная атрофия 5q, фенилкетонурия, адреногенитальный с-м и др.): - действие рецессивного признака проявляется в гомозиготном состоянии; - у здоровых родителей с вероятностью 25% рождаются больные дети, что подтверждает гетерозиготное носительство родителей; - с одинаковой частотой поражаются оба пола; - если болен один из родителей, то, как правило, все потомки рождаются здоровыми, но в 100% гетерозиготными носителями; - в браке между больным и гетерозиготным носителем повторный риск достигает 50% - так называемое "псевдодоминантное наследование", наблюдающееся очень редко. - среди семей с АР формами моногенных болезней (МБ) чаще встречаются кровнородственные браки. Предполагается, что каждый человек в любой популяции является носителем от 5 до 10 рецессивных мутаций, находящихся в гетерозиготном состоянии. |