Рецензенты кандидат химических наук В. Г. Коробко доктор биологических наук В. А. Гвоздев Патрушев Л. И
 Скачать 5.83 Mb. Скачать 5.83 Mb.
|

5.1.2.SOS-мутагенез у бактерийОбразование мутаций в клетках организма, подвергнутого мутагенному воздействию, происходит в основном по одному и тому же механизму. При прохождении репликативного комплекса через некодирующий или ошибочно кодирующий поврежденный участок ДНК наблюдается включение в синтезируемую цепь случайных или соответствующих мутантному участку нуклеотидов. Затем репликация ДНК продолжается в обычном режиме. Таким образом, природа первичных повреждений матричной ДНК определяет тип мутационных замен нуклеотидов в ДНК. У E. coli мутагенез под действием УФ-света и многих химических веществ происходит в результате координированной экспрессии большого числа генов, индуцируемых в ответ на повреждение ДНК. Такая реакция бактериальных клеток на генотоксические воздействия получила название SOS-ответа, а сам процесс образования мутаций – SOS-мутагенеза. В индукции SOS-ответа у E. coli определяющую роль играют два гена: lexA и recA. Белок LexA является репрессором гена recAи более 20 других генов и оперонов, составляющих SOS-регулон. В ответ на повреждение ДНК или ингибирование репликации, как правило, при прохождении ДНК-полимеразой поврежденного участка ДНК вырабатывается внутриклеточный SOS-сигнал. При этом продукт гена recA связывается с одноцепочечными участками ДНК и в результате конформационного перехода обратимо превращается в активированную форму RecA*. Молекулы белка LexA диффундируют к RecA* и взаимодействуют с ним, что сопровождается протеолитическим расщеплением LexA вблизи середины его полипептидной цепи (связь Ala-84Gly-85), что инактивирует LexA как репрессор. В результате происходит индукция LexA-зависимых генов SOS-регулона. В этой реакции белок RecA* не действует как протеиназа, но активирует криптическую протеиназную активность LexA, что завершается расщеплением полипептидной цепи репрессора по аутокаталитическому механизму. Исследование мутантов E. coli, неспособных к SOS-ответу при УФ-повреждениях, привело к открытию еще одного ключевого локуса umuC. Оказалось, что этот локус является опероном, состоящим из двух генов – umuDи umuC. Потеря функции любого из данных генов приводит к подавлению SOS-ответа мутантных бактерий. Таким образом, umuCD-оперон, находящийся под контролем репрессора LexA, входит в состав SOS-регулона, и его функционирование абсолютно необходимо для SOS-мутагенеза. Во время SOS-ответа белок UmuD также расщепляется по аутокаталитическому механизму, запускаемому RecA*. Образующийся в результате полипептид UmuD’ с молекулярной массой 12 кДа, включающий С-концевые остатки UmuD, оказался одним из самых важных компонентов системы SOS-ответа и SOS-мутагенеза у E. coli. Исследования различных мутантных производных UmuD показали, что его нативная форма не является просто неактивным предшественником белка UmuD’, а выполняет функции репрессора SOS-ответа. В растворе UmuD и UmuD’ существуют как в виде гомодимеров, так и более стабильного гетеродимера UmuD–UmuD’, каждый из которых может объединяться с белком UmuС. Комплекс (UmuD’)2–UmuC запускает SOS-ответ, тогда как тримеры (UmuD)2–UmuC и UmuD–UmuD’–UmuC являются в этом отношении неактивными. Образование последнего гетеротримерного комплекса играет важную роль в прекращении клеткой SOS-ответа, так как в этом случае активные внутриклеточные UmuD’ и UmuC выводятся из реакции. Белок RecA, кроме двух вышеупомянутых функций в индукции аутокаталитического расщепления LexA и UmuD, выполняет и третью функцию в SOS-ответе: участвует в формировании так называемых нуклеиново-белковых филаментов в местах одноцепочечных брешей на поврежденной ДНК. С этими филаментами могут соединяться белки UmuD’ и UmuC в составе активного комплекса. Предполагают, что в результате такого взаимодействия комплекс (UmuD’)2–UmuC осуществляет переключение репаративного синтеза ДНК с нужд гомологичной рекомбинации на SOS-мутагенез. Среди других белковых компонентов системы SOS-мутагенеза E. coli необходимо отметить холофермент ДНК-полимеразы III, который проводит включение нуклеотидов в строящуюся цепь ДНК на поврежденном участке. Кроме того, для нормального функционирования системы SOS-мутагенеза необходимы продукты генов groELи groES, являющиеся молекулярными шаперонами. Их роль, как полагают, сводится к стабилизации и обеспечению правильного фолдинга белка UmuC. Таким образом, синтез ДНК на поврежденном участке требует наличия белков UmuD’, UmuC, RecA и холофермента ДНК-полимеразы III. Это было окончательно подтверждено при использовании бесклеточной системы, содержащей все вышеупомянутые белки, а также фрагмент одноцепочечной ДНК с единственным мутантным сайтом без одного азотистого основания. Способность ДНК-полимеразы III преодолевать поврежденный участок ДНК в отсутствие UmuD’, UmuC или RecA была оценена в 0,5%, однако при наличии всех компонентов в реакционной смеси эффективность преодоления поврежденного участка увеличивалась в 10 раз. Очищенная ДНК-полимераза I не заменяет ДНК-полимеразу III в этих опытах. Настоящая роль белкового комплекса (UmuD’)2–UmuC в данном процессе неизвестна. Предполагают, что комплекс может изменять процессивность ДНК-полимеразы, подавлять ее корректирующую 3’→5’-экзонуклеазную активность или изменять конформацию фермента на такую, при которой она начинает неадекватно оценивать пространственную структуру комплекса, образующегося с участием Уотсон–Криковских водородных связей между нуклеотидом матрицы и очередным входящим нуклеотидом. Любое из этих изменений должно сопровождаться повышением частоты ошибочного включения нуклеотидов. Недавно (1998 г.) было установлено, что тример (UmuD’)2–UmuC сам по себе обладает слабой ДНК-полимеразной активностью и, возможно, именно этот комплекс осуществляет синтез ДНК непосредственно в поврежденном участке в присутствии всех вышеупомянутых компонентов. В этой связи тример получил название ДНК-полимеразы V Е. coli. Белок Rev1 дрожжей, гомологичный белку UmuC E. coli, в очищенном состоянии обладает способностью неспецифически включать остатки dCMP в строящуюся цепь ДНК на участке матрицы, в котором отсутствуют азотистые основания. На этом основании он был отнесен к ДНК-полимеразам и был назван дезоксицитидилтрансферазой. Уникальными свойствами обладает новая ДНК-полимераза IV E. coli, кодируемая геном dinB, которая также участвует в SOS-ответе бактерий. Очищенная до состояния, близкого к гомогенному (1999 г.), она не обладает 3'→5'-экзонуклеазной активностью и способна включать нуклеотиды в строящуюся цепь ДНК по высокодистрибутивному механизму. При каждом контакте фермента с субстратом и гибридом праймер–матрица к праймеру присоединяется единственный нуклеотид. В том случае, если вблизи 3'-конца праймера присутствует ошибочно спаренный нуклеотид (особенно пара G–G), то ДНК-полимераза IV в процессе синтеза ДНК осуществляет сдвиг рамки считывания в результате делеции одного нуклеотида. Такой механизм может потенциально исправить мутагенные последствия смещения 3'-конца праймера по отношению к правильной рамке считывания в поврежденной ДНК-матрице. Полагают, что если во время репликации участков ДНК с простыми повторяющимися последовательностями в результате их повреждения происходит включение неправильно спаренного нуклеотида с последующим проскальзыванием 3'-конца строящейся цепи ДНК и образованием мутации со сдвигом рамки считывания, то происходит задержка репликативного комплекса и его диссоциация. В этих условиях синтез ДНК может быть продолжен ДНК-полимеразой IV, которая путем внесения дополнительных мутаций может исправить первоначальный сдвиг рамки. Таким образом, SOS-мутагенез дает возможность микроорганизмам преодолевать летальное действие повреждений ДНК, что особенно важно при нарушениях ее структуры, которые блокируют репликацию ДНК и с которыми не справляется обычная репаративная система. В этом смысле наиболее опасны одноцепочечные бреши, в которых сохранившаяся цепь не содержит азотистых оснований. При таком развитии событий SOS-мутагенез является вторичным процессом – следствием заполнения бреши случайными нуклеотидами. Однако при некоторых видах повреждений во время SOS-ответа в растущую цепь ДНК включаются нуклеотиды, восстанавливающие ее исходную первичную структуру, т.е. происходит истинная репарация повреждений. В этой связи второй функцией SOS-мутагенеза может быть предоставление микроорганизму возможности противостоять определенным генотоксическим воздействиям окружающей среды, а именно таким классам химических мутагенов, последствия действия которых не преодолеваются системами эксцизионной или иной репарации, функционирующими с высокой точностью. Система SOS-мутагенеза, по-видимому, обеспечивает протекание последовательности реакций, обозначаемых как "повторный старт репликации", что имеет место после временной (на 30–45 мин) задержки синтеза ДНК в ответ на ее повреждения генотоксическими агентами. Не исключено также, что SOS-мутагенез создает бактериальным клеткам определенные эволюционные преимущества, так как способствует поддержанию генетического разнообразия в популяциях этих микроорганизмов. 5.1.3.Мутаторный фенотипНесмотря на обилие эндогенных и экзогенных мутагенов, лишь небольшая часть их взаимодействий с ДНК завершается образованием мутаций. Для того чтобы исходное повреждение ДНК в виде аддукта, апуринового сайта или одноцепочечного разрыва закрепилось в геноме в виде мутации, ему необходимо избежать нейтрализующего действия многочисленных ферментов системы репарации ДНК. В экспериментальных условиях для получения требуемых мутаций с помощью химических мутагенов и последующего скрининга требуется большая доза суммарного мутагенного воздействия. Из экспериментальных кривых "доза–эффект" видно, что число возникающих мутаций прямо пропорционально дозе мутагенного воздействия. Исходя из этого уже a priori можно предположить, что повреждение отдельных компонентов системы репарации должно приводить к возрастанию выхода мутаций в ответ на определенную дозу мутагенного воздействия. Действительно, описаны многочисленные штаммы микроорганизмов и линии соматических клеток с повышенными частотами спонтанных мутаций. Совокупность признаков организма, для которой характерна повышенная частота образования спонтанных мутаций, получила название мутаторного фенотипа. Исследование молекулярно-генетических механизмов, приводящих к формированию мутаторного фенотипа, позволило обнаружить отдельные гены, ответственные за этот процесс. Такие гены называют генами-мутаторами, или просто мутаторами. Прежде всего, к ним относятся многие гены системы репарации ДНК, контролирующие разные ее этапы (подробнее см. раздел 5.3). Другие гены, мутации в которых приводят к мутаторному фенотипу, кодируют ферменты матричного синтеза нуклеиновых кислот. Описаны мутационные замены отдельных аминокислотных остатков в ДНК-полимеразах, которые понижают специфичность выбора дезоксирибонуклеозидтрифосфатов из внутриклеточного пула в соответствии с последовательностью матричной ДНК. Следствием этого является повышение частоты включения некомплементарных матрице нуклеотидов в строящиеся цепи ДНК. Однако сам процесс включения некомплементарных матрице нуклеотидов является лишь одной из стадий, критических для контроля точности репликации ДНК. Благодаря наличию у ДНК-полимераз корректирующей 3’→5’-экзонуклеазной активности ошибочно включенные некомплементарные матрице нуклеотиды тотчас удаляются из строящейся цепи ДНК, что защищает ее от точковых мутаций, возникающих по такому механизму. Поэтому неудивительно, что мутации, нарушающие функционирование корректирующей экзонуклеазной активности, также приводят к возникновению мутаторного фенотипа. К аналогичным эффектам приводят нарушения функционирования систем рекомбинации, транскрипции, систем контроля структуры хроматина, ферментных систем, контролирующих сегрегацию хромосом и число копий индивидуальных генов, а также систем, участвующих в синтезе эндогенных мутагенов. Нарушения функционирования и координации экспрессии генов метаболизма нуклеотидов также приводят к мутаторному фенотипу. Известно, что повышение внутриклеточной концентрации дезоксирибонуклеозидтрифосфатов сверх оптимального уровня понижает точность репликации ДНК. Мутаторный фенотип после возникновения начинает имитировать непрерывное мутагенное воздействие, интенсивность которого зависит от характера повреждений генов-мутаторов. Мутаторный фенотип у микроорганизмов проявляется в повышенной частоте возникновения спонтанных мутаций, которые можно измерить по частоте появления клеток, выживающих в селективных условиях, например в присутствии антибиотиков. У эукариот мутаторный фенотип часто сопровождается дестабилизацией генома, обнаруживаемой по возрастанию частоты внутригеномных перестроек ДНК. В связи с этим явлением наиболее интенсивно исследуются изменения структуры микросателлитных повторов в геноме человека при онкологических заболеваниях. Сравнение структуры отдельных микросателлитных локусов в клетках опухолей и нормальных тканей одного индивидуума часто обнаруживает существенные различия между микросателлитами одного и того же локуса. Такого рода исследования чаще всего проводятся с помощью ПЦР или любого другого метода, используемого при ДНК-типировании (см. главу 10). 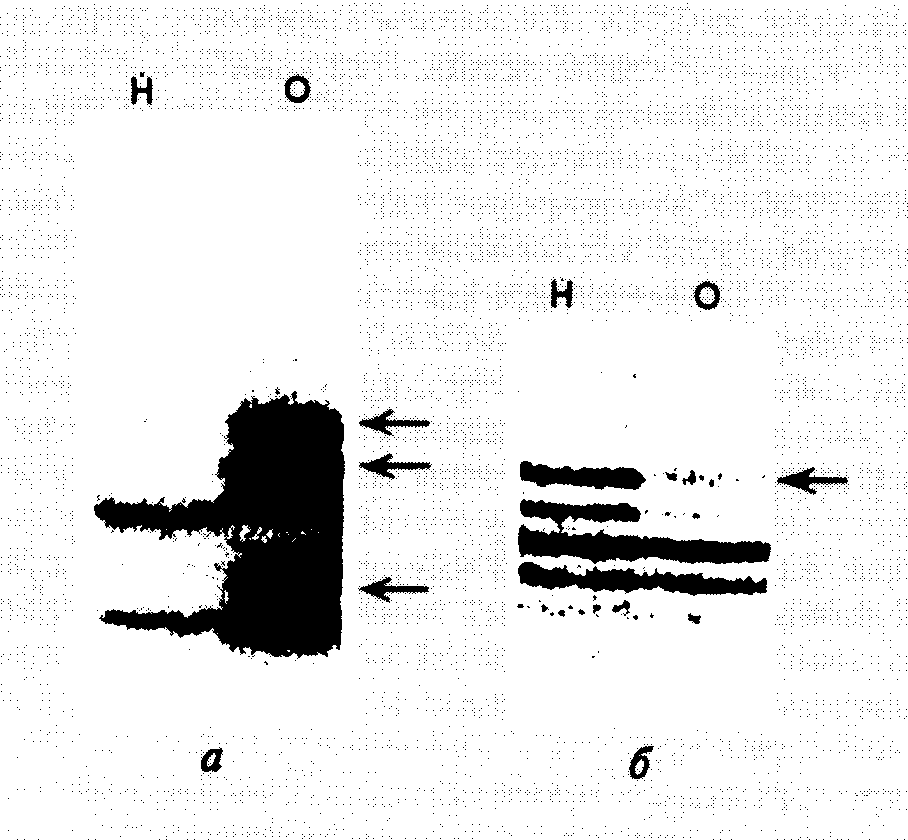 Рис. I.54. Примеры нестабильности микросателлитов (а, на примере карциномы молочной железы) и потери гетерозиготности (б, на примере глиомы) при различных онкологических заболеваниях. Показаны продукты ПЦР после разделения с помощью электрофореза в полиакриламидном геле. Н – нормальная ткань, О – опухоль. Стрелки указывают на измененные аллели Наиболее просто обнаруживаются два типа изменений микросателлитов при мутаторном фенотипе (рис. I.54). При одном из них изменение суммарной длины микросателлитных повторов конкретного генетического локуса обнаруживают по возрастанию или уменьшению электрофоретической подвижности соответствующих продуктов ПЦР при сравнении его состояния в опухолевых и нормальных тканях одного и того же организма (см. рис. I.54,а). В исследованиях подобного рода часто наблюдают эффект так называемой потери гетерозиготности исследуемых микросателлитных локусов. Ввиду диплоидности генома человека каждый микросателлитный локус в нем представлен двумя копиями и, следовательно, двумя аллелями в случае его гетерозиготности. При ее потере происходит выравнивание длины обоих микросателлитных аллелей, которые различаются в нормальных тканях, или удаление одного из аллелей в результате делеции. Удаление фиксируют электрофоретически по исчезновению одного из продуктов ПЦР после амплификации соответствующих локусов, не сопровождаемому появлением новых полос (см. рис. I.54,б). Дестабилизация микросателлитных локусов в опухолевых клетках, по-видимому, не служит непосредственной причиной малигнизации этих клеток. Нестабильность микросателлитов скорее может быть чувствительным маркером мутаторного фенотипа раковых клеток, внешним проявлением дестабилизации генома, характерного для опухолевых клеток многих типов. Варьирование размеров микросателлитных локусов является частным случаем большой группы мутаций, связанных с изменением числа копий последовательностей нуклеотидов в геноме эукариот. В качестве еще одного примера мутаций этого рода рассмотрим изменения размеров небольших кластеров ди- и тринуклеотидных повторов в геномной ДНК. Недавно было установлено, что такие мутации, получившие название экспансии ДНК, лежат в основе многих тяжелых заболеваний человека. 5.1.4.Экспансия ДНКПод экспансией ДНК понимают увеличение числа копий коротких повторяющихся последовательностей нуклеотидов внутри кластера при передаче генетической информации от родителей потомкам. В настоящее время различают два класса этих генетических явлений. При экспансии ДНК первого класса происходит резкое и стабильное увеличение числа копий определенных повторов (10) на фоне полного отсутствия обратного сокращения длин их кластеров. При экспансии ДНК второго класса изменения затрагивают меньшее число повторов (4), а образование мутационных вставок и обратных делеций повторяющихся последовательностей происходит с одинаковой скоростью. Мутации первого класса ассоциируются с болезнью Хантингтона, синдромом ломкости X-хромосомы, болезнью Кеннеди, миотонической дистрофией, спиноцеребральной атаксией типа I и рядом других неврологических заболеваний. У здоровых индивидуумов имеет место полиморфизм по числу копий повторяющихся единиц в этих локусах, и экспансия повторов приводит к развитию таких заболеваний после того, как количество копий повторяющихся единиц (как правило, тринуклеотидов) начинает превышать определенное значение. В результате изменяется уровень экспрессии гена, содержащего повторы, или же свойства кодируемого геном белка. Экспансия динуклеотидов CA или AT, относящаяся ко второму классу мутаций, ассоциируется чаще всего с наследственным раком кишечника. В наследовании предрасположенности к экспансии ДНК имеет место импринтинг (см. раздел 3.2.5). Процесс экспансии происходит во время митотической репликации ДНК на ранних стадиях эмбриогенеза. Для развития синдрома ломкости X-хромосомы только в материнской X-хромосоме должен образовываться премутационный аллель, содержащий 50–200 копий тринуклеотида CGG. При наличии такой премутации в эмбриональном развитии мужской особи формируется мутантный аллель, содержащий 200–2000 копий этого повтора. Подобной экспансии ДНК никогда не происходит, если премутация локализована на отцовской X-хромосоме. Приведенный пример с синдромом ломкости X-хромосомы указывает на необходимое условие для экспансии ДНК и во всех остальных случаях: увеличение числа копий повторов у родителей до определенного порогового значения. Как только формируется премутантный аллель в геноме родителей, у следующего поколения происходит образование истинно мутантного аллеля, сопровождаемое развитием соответствующего патологического состояния организма. 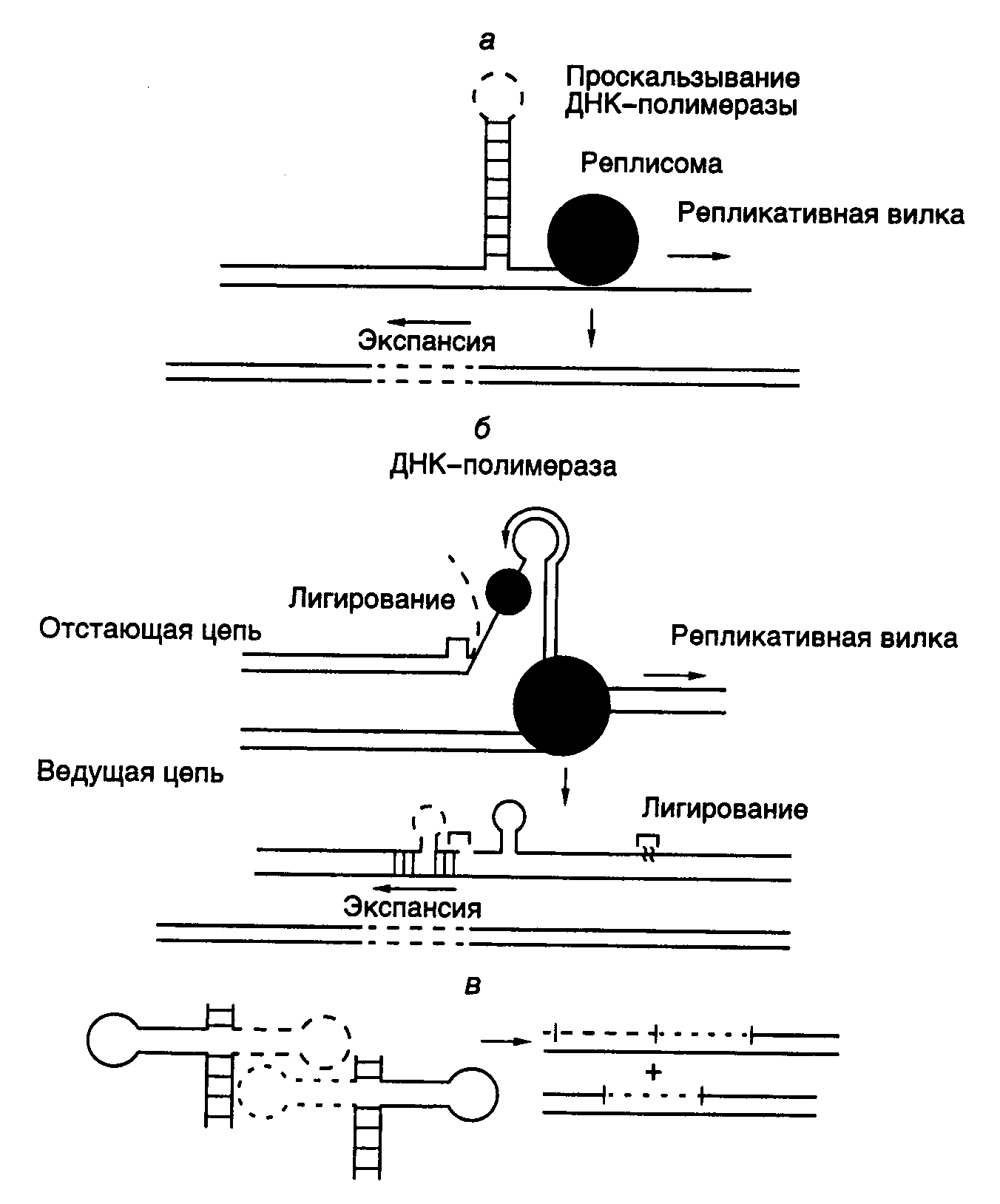 Рис. I.55. Модели механизма экспансии ДНК во время репликации а – с проскальзыванием ДНК-полимеразы; б – с участием отстающей цепи ДНК; в – с привлечением механизма конверсии генов Среди возможных ди- и тринуклеотидных последовательностей лишь для CAG/CTG, CGG/CCG и AT продемонстрирована экспансия in vivo, приводящая к патологиям. Предполагают, что возможность экспансии именно этих, а не других повторов определяется их способностью образовывать стабильные шпильки. Предложены три модели, объясняющие механизм экспансии ДНК, в которых участвуют шпильки, образованные соответствующими повторами (рис. I.55). Первая модель предполагает проскальзывание реплицирующей молекулы ДНК-полимеразы на повторе в обратном направлении, сопровождаемое образованием шпильки в строящейся цепи ДНК (см. рис. I.55,а). В этом случае в следующем раунде репликации или по завершении репаративного синтеза ДНК длина кластера повторяющихся последовательностей увеличится на размер сегмента ДНК, образовавшего шпильку. Согласно второй модели при репликации отстающей цепи ДНК на соответствующем повторе матричная ДНК некоторое время находится в одноцепочечной форме (см. рис. I.55,б). В это время одноцепочечная последовательность отстающей цепи, содержащая повтор, может образовать шпильку, которая блокирует синтез ДНК в данном месте. Предполагается, что белок, связывающийся с одноцепочечной ДНК (SSB-белок), не может эффективно взаимодействовать с двухцепочечной шпилькой, однако сохраняет способность связываться с одноцепочечной петлей на вершине шпильки, обеспечивая инициацию репликации ДНК с этого места. После того как ДНК-полимераза прореплицирует одну половину шпильки, ее структура разрушится, что приведет к снятию блока репликации перед шпилькой и ее нормальному продолжению. Однако синтезированный перед этим фрагмент ДНК, соответствующий одной половине шпильки, оказывается лигированным с уже имевшимся фрагментом Оказаки. Он не может отделиться от матрицы, которая своими нуклеотидами, расположенными по концам, образует водородные связи с повторами матрицы. Далее избыточная последовательность включается в растущую цепь ДНК и после ошибочной репаративной коррекции шпильки приводит к увеличению размера этого кластера повторяющихся последовательностей, т.е. их экспансии. В соответствии с третьей моделью шпильки, образующиеся в местах соответствующих кластеров повторяющихся последовательностей на разных молекулах ДНК, могут взаимодействовать друг с другом, что будет сопровождаться возникновением сложно структурированной матрицы для репликации. В процессе синтеза ДНК на такой сложной матрице весьма вероятно включение избыточной последовательности в виде повтора во вновь синтезируемую ДНК (см. рис. I.55,в). Аналогичный эффект может быть достигнут и в результате сложной последовательности событий рекомбинации с участием этих шпилек. Заканчивая обсуждение недавно обнаруженных "необычных" мутационных изменений геномной ДНК, связанных с увеличением числа копий (экспансией) коротких повторяющихся последовательностей, следует подчеркнуть, что они не изменяют традиционных представлений о мутагенезе. Хотя такие мутации ассоциированы с конкретными генетическими локусами, содержащими повторы определенной структуры, процесс экспансии этих последовательностей носит случайный характер и, возможно, запускается первичным мутационным изменением в кластере повторов, которое делает более вероятным формирование шпилечной или иной пространственной структуры подобных кластеров. Однако не так давно была описана еще одна группа мутаций, само существование которых идет в разрез с общепринятым неодарвиновским представлением о механизмах возникновения мутаций. В следующем разделе речь пойдет об адаптивных мутациях; их возникновение в геноме носит не случайный, а направленный характер. 5.1.5.Адаптивные мутацииПроблема, связанная с возможностью возникновения адаптивных мутаций, имеет глубокие корни в биологии. За 50 лет до того как Ч. Дарвин начал свои знаменитые исследования происхождения биологических видов, другой биолог, француз Ж.Б. Ламарк, разработал учение о возможности наследования признаков, которые родительские организмы приобретали на протяжении жизни под действием окружающей среды. В терминах современной генетики это означает, что организм в ответ на определенное воздействие внешних факторов может целенаправленно изменять свой геном в клетках зародышевой линии таким образом, что он у потомков будет контролировать развитие признаков, максимально адаптированных к изменившимся внешним условиям. Следовательно, по Ламарку организм самостоятельно способен делать выбор между полезными и вредными признаками и тем самым направлять свои эволюционные преобразования. Поскольку любое наследуемое изменение генома представляет собой мутацию, неоламаркистские концепции предполагают, что организм может контролировать мутационные изменения своего генома и направлять их в нужное русло развития признаков, полезных для выживания. Взгляды неодарвинистов на мутационный процесс прямо противоположны. В соответствии с их доктриной, доминирующей в системе взглядов современных молекулярных генетиков, мутации являются случайными и спонтанными событиями, а образующиеся мутантные признаки подвержены жесткому давлению естественного отбора. В популяциях закрепляются лишь мутации (и признаки), максимально соответствующие условиям окружающей среды. Остальные элиминируются в результате гибели мутантных особей. Считается, что спор между неоламаркистами и неодарвинистами окончательно решен в пользу последних, по крайней мере, тремя классическими экспериментами. Во флуктуационном тесте (С. Лурия, М. Дельбрюк, 1943 г.) исследовали возникновение мутантов E. coli, устойчивых к бактериофагу Т1 в независимо выращиваемых культурах бактерий. Бактериальные клетки из разных пробирок высевали на чашки Петри с избытком фага Т1 и подсчитывали число образующихся бактериальных колоний, устойчивых к бактериофагу. Предполагалось, что если мутантные бактерии образуются в пробирках до вступления в контакт с бактериофагом, то количество устойчивых бактерий будет сильно различаться в разных пробирках в зависимости от числа делений, которые совершит мутантная бактерия с момента возникновения мутации до высева на чашку Петри. В том случае, когда мутации возникают после взаимодействия бактерий с вирусом, количество мутантных бактерий, обнаруживаемых на разных чашках, будут следовать непрерывному распределению Пуассона. В ходе этих экспериментов продемонстрировано предсуществование мутантных бактерий в культурах. В еще более наглядных опытах Дж. Ледерберга и Е.М. Ледерберг (1950 г.) с помощью бархатного штампа перепечатывали бактерии с газона на разные чашки Петри, содержащие бактериофаг Т1. Оказалось, что мутантные бактерии, устойчивые к бактериофагу, образуют колонии в одних и тех же местах разных чашек. Эти данные были также в пользу предсуществования мутантных бактерий в газоне, не имевшем контакта с бактериофагом. Однако в своих первых опытах исследователи имели дело с потомками исходных бактериальных клеток, которые контактировали с селектирующим агентом (бактериофагом Т1) в момент отбора. Поэтому "окончательное" решение вопроса было достигнуто после получения в 1956 г. штаммов бактерий, устойчивых к стрептомицину и ранее не соприкасавшихся с антибиотиком. Потрясение твердо устоявшихся основ молекулярной генетики с неоламаркистских позиций началось в 1988 г. после опубликования в журнале "Nature" статьи Дж. Кэрнса, Дж. Овербаха и С. Миллера "Происхождение мутантов". В серии экспериментов с мутантными клетками E. coli, неспособными использовать лактозу в качестве источника углерода (фенотип Lac-), авторы установили, что скорость образования ревертантов в том случае, если мутантные бактерии инкубировали на чашках в присутствии лактозы, значительно превышала ожидаемую из случайного возникновения обратных мутаций в стационарной бактериальной культуре. На этом основании авторы сделали вывод о том, что селективные условия (присутствие неусваиваемой лактозы в качестве единственного источника углерода) оказывают влияние на спектр мутаций, возникающих у бактериальных клеток. В работе утверждается, что бактериальные клетки могут сами контролировать свой мутационный процесс, направляя его в сторону образования нужных мутантных ферментов, что позволяет клеткам адекватно реагировать на сигналы окружающей среды, которая направленно формирует генотип бактериальных клеток. Такие "еретические" выводы Кэрнса и его соавторов получили экспериментальное подтверждение в его дальнейших исследованиях, а также в многочисленных работах других авторов с использованием бактериальных и дрожжевых клеток в качестве объекта. И хотя в ряде случаев было показано наличие артефактов, приводивших к неправильной интерпретации результатов, в целом существование феномена направленного образования адаптивных мутаций подтверждено и пока не опровергнуто. Однако он может занять достойное место среди других хорошо доказанных генетических явлений лишь после экспериментального выяснения молекулярных механизмов, лежащих в основе адаптивных мутаций. Для объяснения этих фактов в настоящее время выдвинуто несколько гипотез, ни одна из которых пока не получила полного экспериментального подтверждения. По мнению Кэрнса и соавторов (1988 г.) клетки синтезируют набор вариабельных, незначительно различающихся по первичной структуре молекул мРНК, и путем обратной транскрипции получают копию кДНК с одной из них, кодирующей наиболее подходящую для адаптации белковую молекулу. Далее такая кДНК в результате рекомбинации замещает мутантный аллель в геноме микроорганизма. Б.Д. Дэвис (1989 г.) считает, что индукция транскрипции отдельных локусов в геноме покоящихся микроорганизмов, в частности лактозой, повышает их мутабильность. Ф.У. Сталь (1988 г.) и Л. Боэ (1990 г.) высказывают предположение о снижении функционирования систем репарации ДНК у голодающих микроорганизмов, что может быть причиной повышения частоты мутаций в транскрибируемых локусах. Те же авторы предполагают, что в основе феномена направленного повышения частоты мутаций лежит recA-зависимая амплификация соответствующих генетических локусов, сопровождаемая корректирующим мутагенезом. Б.Г. Холл (1990 г.) для объяснения адаптивных мутаций разработал модель, в соответствии с которой в популяции голодающих микроорганизмов часть клеток находится в состоянии повышенной мутабильности. Среди этих клеток выживают лишь мутанты, максимально соответствующие требованиям окружающей среды. По крайней мере, два результата исследований последних лет делают концепцию Холла наиболее приемлемой. Прежде всего было установлено, что реверсия мутантных бактериальных клеток к фенотипу Lac+ в условиях голодания требует функционирования RecBCD-зависимой репарационной системы рекомбинации (см. раздел 5.2.3). Кроме того, в бактериальном геноме были обнаружены горячие и холодные точки, в которых образование адаптивных мутаций может происходить соответственно с высокой и низкой частотой, что объясняет отмеченную в литературе невозможность их обнаружения в некоторых генетических локусах. Поскольку на первых этапах работы репарационной системы рекомбинации происходит внесение в ДНК двухцепочечных разрывов, с которыми далее взаимодействует комплекс белков RecBCD, полагают, что такие разрывы ДНК инициируют процесс возникновения адаптивных мутаций. Во время рекомбинационного обмена цепями ДНК, индуцированного двухцепочечными разрывами, происходит синтез новых цепей ДНК-полимеразой III, сопровождаемый ошибочным включением нуклеотидов. (Как уже упоминалось в разделе 5.1.2, ДНК-полимераза III является активным участником SOS-мутагенеза у бактерий.) Такие ошибочно включенные нуклеотиды с высокой вероятностью могут закрепляться в геноме в виде мутаций из-за ослабления эффективности функционирования системы эксцизионной репарации у бактериальных клеток, находящихся в стационарной фазе роста, и, следовательно, формирования у голодающих бактериальных клеток мутаторного фенотипа. При случайном возникновении мутации, возвращающей клетку к нормальному Lac+-фенотипу, мутантная бактерия выходит из стационарной фазы и начинает активно делиться. При этом происходит восстановление обычного функционирования системы репарации. Как можно видеть, обсуждаемая модель не оставляет места классическому неоламаркизму. При реализации такого механизма возникновение "адаптивной" мутации определяет случай, и после ее появления происходит клональное замещение исходной популяции бактерий мутантными клетками. Однако выбор самих генетических локусов, в которых могут происходить такие мутации, уже не является случайным. Он генетически детерминирован, на мой взгляд, структурой бактериального генома. В соответствии с развиваемой в разделе 5.3 концепцией альтруистичной ДНК, адаптивные мутации являются в конечном итоге естественным следствием дифференциальной защиты отдельных генетических локусов от спонтанных мутаций, определяемой пространственной структурой ДНК локусов. Как будет следовать из дальнейшего изложения, частота мутаций в первом приближении обратно пропорциональна уровню конденсации ДНК конкретного генетического локуса и определяется физической доступностью отдельных его частей химическим мутагенам и(или) ферментам системы репарации. Кроме того, индукция транскрипции этих предетерминированных локусов или даже простое удаление регуляторных белков из промоторной зоны генов могли бы изменять их пространственную структуру и, как следствие, уровень мутабильности соответствующих участков генома. 5.1.6.Механизмы защиты генома от мутацийНесмотря на то что иногда мутации помогают организму выжить, подавляющее большинство мутационных изменений генома нежелательно и сопровождается развитием различных патологических состояний мутантной особи или отдельной соматической клетки. Жестко действующий естественный отбор, в частности, через систему иммунного надзора элиминирует мутантные соматические клетки, опасные для существования многоклеточного организма, например предотвращая иногда развитие онкологических или аутоиммунных заболеваний. Однако к гораздо более плачевным последствиям приводит элиминация естественным отбором целой мутантной особи, так как это сопровождается непродуктивной гибелью большого числа соматических клеток и является расточительным с точки зрения энергетических затрат на их биосинтез. Генетическая информация любого организма тщательно защищена от мутационных повреждений, что делает мутации в жизненно важных локусах генома очень редкими. Защита осуществляется на нескольких уровнях. Прежде всего, организм старается не допустить попадания химических мутагенов в жизненно важные локусы своего генома. Это достигается двумя путями. Во-первых, избыточные последовательности нуклеотидов ДНК, экранируя кодирующие последовательности нуклеотидов в геноме эукариот, принимают удар большей части химических мутагенов на себя, не допуская их попадания в такие локусы. Те же цели могут быть достигнуты за счет особой пространственной организации ДНК в конкретных участках генома (подробнее см. раздел 5.3). Во-вторых, в клетках имеются многочисленные высоко- и низкомолекулярные ловушки мутагенов, важнейшими из которых являются: маннит, энкефалины, индолы, желчные кислоты и их производные, -токоферол, аскорбиновая кислота, тирозин, серотонин, а также ряд других соединений экзогенного и эндогенного происхождения. К сожалению, обе системы защиты не обладают 100%-й эффективностью. То же можно сказать и о точности функционирования ферментных систем, осуществляющих воспроизведение генетической информации. Поэтому многочисленные нарушения первичной структуры ДНК неизбежны. Тем не менее, большинство первичных повреждений не превращается в мутации благодаря функционированию высокоэффективных систем репарации ДНК, состоящих из многих ферментных компонентов. Из-за исключительной важности функционирования систем защиты генетической информации в поддержании эффективной экспрессии генов ниже будут рассмотрены основные компоненты систем репарации ДНК и принципы их работы. |