Решение перечисленных задач возможно только при наличии достаточного количества зарегистрированных и доступных для измерения показателей, отражающих уровень качества продукции.
 Скачать 253.46 Kb. Скачать 253.46 Kb.
|

  Сигнал детектора А + В    В В Д А + В Д А + В  В А А + В В А А + В  А + В А В А + В А В    Время А IV. В зависимости от способа оформления процесса различают колоночную хроматографию и плоскую (на бумаге и тонкослойную) В колоночной хроматографии — неподвижная фаза в виде мелкозернистого твёрдого адсорбента или инертного материала, пропитанного определённой жидкостью, находится в длинной узкой трубке. Такая колонка называется насадочной. Применяются также капиллярные колонки, в которых неподвижная фаза покрывает тонким слоем внутреннюю стенку колонки. В тонкослойной хроматографии используются стеклянные пластинки, на которых нанесен тонкий слой сорбента, а также листы специальной хроматографической бумаги. V. По назначению хроматографию подразделяют на: аналитическую — для проведения качественного и количественного анализа препаративную — для выделения небольших количеств чистых веществ промышленную — для получения чистых веществ в больших количествах 5.1.2 Теоретические основы хроматографии. Понятие о хроматограмме. Расшифровка хроматограмм (расчёт на основе её параметров). Задачей теории хроматографии является установление закона движения хроматографических зон и выбор оптимальных условий разделения. Хроматографическое разделение основано на различной сорбируемости компонентов. Сорбируемость описывается изотермой сорбции, отражающей зависимость концентрации в неподвижной фазе (сорбенте) — Са от концентрации в подвижной фазе — С. С  а а  С Линейные участки изотерм, соответствующие малым концентрациям линейны и описываются законом Генри. Са = К · С К — константа Генри, она характеризует сорбируемость данного компонента, чем больше К, тем больше сорбируемость. Разделение определяемого компонента между фазами. В любой хроматографической системе происходит обратимый переход молекул определяемого компонента А из подвижной фазы (ПФ) в неподвижную (НФ), при этом: Сп.ф. ↔ Сн.ф. (устанавливается равновесие) Сорбируемый процесс в хроматографической колонке характеризуется константой равновесного распределения или коэффициентом равновесного распределения, который представляет собой отношение равновесной концентрации вещества в неподвижной фазе к концентрации вещества в подвижной фазе: Краспр. = [Сн.ф.] / [Сп.ф.] Рассмотрим Краспр. для компонента А: Краспр. = [Ан.ф.] / [Ап.ф.] [Ан.ф.] = mАн.ф. / Vн.ф. [Ап.ф.] = mАн.ф. / Vп.ф. отсюда: Краспр. = 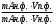 k` = k` =  Краспр. = k` Краспр. = k` 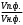 где: mАн.ф., mАп.ф. — количество компонента А в неподвижной и подвижной фазах Vп.ф., Vн.ф. — объёмы подвижной и неподвижной фазы K — коэффициент ёмкости Коэффициент распределения зависит от природы определяемого компонента, природы подвижной и неподвижной фазы, температуры, рН, концентрации и др. скорость движения зоны данного вещества обратно пропорциональна коэффициенту распределения Краспр. При больших значениях Краспр. — большая часть определяемого компонента находится в неподвижной фазе (НФ) и перемещается медленно. Если Краспр. малая величина, то определяемый компонент быстро продвигается по колонке. Два компонента с различными Краспр. будут перемещаться с разными скоростями, что является определяющим фактором хроматографического разделения. При хроматографических определениях вещество подвижной фазы (ПФ) вступает в контакт с участками неподвижной фазы (НФ) — сорбента, проходит через весь его слой и на выходе из колонки поток подвижной фазы — носителя вступает в детектор, который регистрирует изменение свойств потока, изменения эти происходят за счёт изменения концентрации определяемого компонента в потоке. Понятие о хроматограмме. Вещество подвижной фазы, содержащее жидкий или газообразный носитель и определяемые компоненты вступает в контакт с участками неподвижной фазы — сорбента, проходит весь его слой и попадает в детектор, который регистрирует изменение свойств потока, эти изменения происходят за счёт изменения концентрации определяемого компонента в потоке. Это приводит к изменению сигналов детектора, фиксируемых лентой самописца. Эта запись, как уже упоминалось, называется хроматограммой. Хроматограмму называют также выходной кривой. Она служит выражением результатов хроматографического разделения веществ. Сорбционная способность неподвижной фазы характеризуется временем удерживания (tR) или объёмом удерживания (VR), который представляет объём подвижной фазы, прошедшей через слой сорбента за время tR. Между ними зависимость υR = tR · υ υ — объёмная скорость подвижной фазы Нулевая линия — часть хроматограммы, полученная при регистрации сигнала детектора во время выхода из колонки чистой подвижной фазы. Существуют другие параметры хроматограммы С    tR2 tR2 tR1 tR1  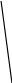 ΔtR2,1 ΔtR2,1h  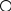 3 4 3 4  tR0 tR0   2 1 2 1    μ μ 0,5 μ μ 0,5    V 1 — нулевая линия; 2 — пик несорбируемости компонента; 3, 4 — пики определяемых компонентов. Высота выходной кривой — высота пика h — перпендикуляр из максимума пика на нулевую линию. Ширина пика μ — отрезок отсекаемый на нулевой касательной к кривой в точке перегиба (или на половине высоты — μ0,5). tR — промежуток времени от момента ввода пробы до достижения max h на пике. Расшифровка хроматограммы. Расшифровка хроматограммы сводится к определению высоты и ширины пика, если пик узкий, то определяют только высоту пика. Высота пика определяется как высота перпендикуляра, проходящего через max точку пика на нулевую точку пика, если пик пологий, то высоту проводят из точки пересечения касательных. Для широких пиков определяется не только высота, но и ширина, т.к. устанавливается зависимость между концентрацией компонента и высотой и площадью пика.      h h h h 1  /2h h /2h h    a a a a h = fC S = fC S = h · ½ a — основание Ширина пика находится как ширина основания треугольника, полученного при пересечении двух касательных с нулевой линией. (иногда используют 1/2h). Хроматографический метод анализа можно использовать как для качественного, так и для количественного анализа. 1) В качественном анализе используют несколько методик расшифровки хроматограмм. а) качественный анализ по времени удержания Rуд. (τуд.) Rуд. (τуд.) — время удержания – это промежуток времени от момента ввода пробы до выхода max на хроматограмме, оно зависит от условий хроматографирования, от природы анализируемого компонента. Метод заключается в том, что отмечают τуд. эталонной смеси, затем исследуемой. При сравнении судят о составе смеси. б) качественный анализ по форме хроматограмм. Вначале получают хроматограмму исследуемой смеси, затем вводят в анализируемую смесь предполагаемое вещество. Если введённое вещество в смеси есть, то увеличивается величина соответствующего пика, если отсутствует — появляется дополнительный пик.   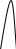 а) б) в) а) б) в)   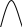 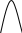 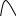 Хроматограмма в-ва нет есть (первичная) Если в анализируемую смесь, имеющую хроматограмму (а) ввести этиловый спирт и прохроматографировать, то может быть два новых вида хроматограмм. (б) — говорит об отсутствии этилового спирта (в) — говорит о присутствии этилового спирта в) табличный качественный анализ с применением хроматографирования исследуемого и введённого эталонного раствора. На основании полученных хроматограмм рассчитывают относительно удерживаемые объёмы по времени удержания и сравнивают с табличными данными и по ним проводят идентификацию: Vотн. = (τуд.иссп. – τ0) / (τуд.эт. – τ0) Где: τуд.иссп — время удержания исследуемой смеси τуд.эт — время удержания эталона τ0 — время удержания газа-носителя Иногда используют не время удержания, а расстояние в мм (i) от момента вкалывания пробы до появления max пика. Vотн. = (iуд.иссп. – τ0) / (iуд.эт. – τ0) 2) Количественный анализ основан на методиках, учитывающих изменение различных параметров пика, зависящих от концентрации анализируемых компонентов — h, a, S и VR или hVR. а) Метод нормировки — сумму параметров пиков (h, S) принимают за 100 % и массовая доля находится как отношение h и S отдельных пиков к этой сумме (х100) % = 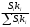 · 100 % = · 100 % = 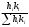 · 100 · 100б) Метод абсолютной калибровки — наиболее точен. В нём экспериментально определяют зависимость h или S от концентрации и строят калибровочные графики по стандартным растворам, а потом хроматографируют смесь и по высоте полученных пиков определяют С. Если тщательно готовить стандарт смеси и выдерживать условия хроматографирования, метод отличается высокой точностью. в) Метод внутреннего стандарта основан на введении в анализируемую смесь точно известного количества стандарта, близкого по физическим свойствам к компонентам смеси. Смесь хроматографируют, определяют h и S. % = 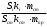 · 100 · 100Дать пример расчёта на метод нормировки и внутреннего стандарта. Примеры расчётов. 1. При хроматографировании смеси компонентов, расшифровка хроматограммы дала следующие данные:
Определить %-ное содержание компонентов в смеси, используя метод нормировки. 2. Вычислить % толуола в пробе по методу внутреннего стандарта, если данные хроматографирования: (в качестве внутреннего стандарта – бензол) 3. При определении бутилового спирта методом газовой хроматографии были получены следующие пики в зависимости от содержания, используя калибровочный график:
Для 0,02 мл исследуемого раствора получили пик h = 57 мм. Определить %-ное содержание спирта, если ρ = 0,91 г/мл 3). Методы идентификации в газовой хроматографии. В газовой хроматографии параметры удерживания какого-либо соединения в смеси при определённых условиях характеризуют природу этого соединения, поэтому они (параметры удерживания) могут быть использованы для целей идентификации. В качестве параметров для идентификации чаще всего используют время удерживания tR, удерживаемый объём VR, логарифмический индекс удерживания J. В практике качественного газохроматографического анализа используют следующие способы идентификации компонентов: Сравнение параметров удерживания неизвестного вещества и эталонного соединения при идентичных условиях хроматографирования. Применение графических или аналитических зависимостей между характеристиками удерживания и физико-химическими свойствами веществ (молекулярной массой, tкип., числом углеродных атомов или функциональных групп и т.д.). Сочетание газовой хроматографии с другими инструментальными методами (ИК-спектроскопией и др.). Применение селективных детекторов. 4). Практическое применение. Большое значение газовой хроматографии в практике вызвано тем, что с её помощью можно идентифицировать отдельные компоненты сложных газовых смесей и определить их количество. Метод является универсальным и не требует больших затрат времени. Этим методом анализируют нефтяные и рудничные газы, воздух, продукцию основной химии и промышленности органического синтеза, нефть и продукты её переработки, производят разделение изотопов некоторых изотопов. Хроматография широко используется в биологии и медицине, в технологии переработки древесины, в лесохимии, пищевой промышленности и многих других. Методы газовой хроматографии в физико-химических исследованиях, для анализа сложных многокомпонентных систем, определение микропримесей, а также для определения защитных свойств противогазных коробок и фильтр-поглощающих элементов. 5.1.3 Газовая хроматография (ГХ). Её виды Подвижная фаза — газ или пар (газ-носитель). В зависимости от состояния неподвижной фазы различают газо-адсорбционную (ГХ) и газо-жидкостную (ГЖХ) хроматографию. В ГХ — неподвижной фазой является твёрдый адсорбент. В ГЖХ — неподвижной фазой является жидкость, плёнка жидкости на поверхности частиц твёрдого сорбента. Газовая хроматография основана на различной сорбируемости компонентов смеси, применима для анализа смеси газов, легколетучих жидкостей и некоторых твёрдых веществ, способных переходить в паро- или газообразное состояние. В качестве газа-носителя используют инертные газы — Не, Ne, Ar, а также N2, H2, CO2 и др. Скорость газа-носителя поддерживают постоянной. Требования к газу-носителю: Должен быть инертен по отношению к определяемым компонентам. Должен быть химически чистым. Быть дешёвым и легкодоступным. Подходить к детектору. В ГХ колонки заполняются твёрдым сорбентом. В качестве сорбентов может применяться активированный уголь, графит, силикагель, оксид алюминия, цеолиты и т.д. Активированные угли неполярны, обладают высокой удельной поверхностью 1000-1700 м2/г, что обуславливает большую силу взаимодействия с анализируемым веществом. Силикагель и оксид алюминия — полярные адсорбенты, на их поверхности имеются заряды. Применяемые в качестве сорбента цеолиты, являются алюмосиликатами щелочных металлов. Их можно рассматривать как молекулярные сита, т.к. их поры имеют размеры, близкие к размерам молекул и адсорбция на них является своеобразным “просеиванием”, сорбируются, в основном, вещества, молекулы которых могут проникать внутрь кристаллической решётки. Сравнительно недавно начали использовать полимерные сорбенты на основе сополимеров стирола, этилстирола, дивинилбензола и др. Требования к сорбенту: Должен быть однородным. Должен иметь большую поверхность. Не должен взаимодействовать ни с компонентами смеси, ни с газом-носителем. Обладать активностью. Не иметь каталитических свойств. При проведении работ по методу газовой хроматографии в колонке происходит процесс адсорбции газа на твёрдом адсорбенте, при использовании газожидкостной хроматографии вместо процесса адсорбции — стал происходить процесс растворения газа в тонкой плёнке, находящейся на твёрдом носителе и эффективность разделения стала определяться не процессами адсорбции — десорбции газа, как это происходит в адсорбционной газовой хроматографии, а процессами растворения газа в жидкой плёнке и его выделения. Дозаторы — устройства, предназначенные для ввода пробы. Проба может быть введена непосредственно в поток газа-носителя или в специальный дозирующий объём. Небольшие количества вводят с помощью специальных микрошприцев (или медицинских). Большие по объёму пробы вводят с помощью газовых пипеток, твёрдые пробы растворяют и вводят в виде раствора с помощью микрошприца. Принципиальная схема газового хроматографа |