Системы крови
 Скачать 7.82 Mb. Скачать 7.82 Mb.
|

|
Рис. 26-4. Активация протеина С и его антикоагулянтная активность В механизме фибринолитической активности крови имеет значение инактивация ингибитора активатора плазминогена первого типа, образующегося в эндотелии. Таким образом, активированная тромбином система ПрС-ПрS, с одной стороны, ингибирует гемокоагуляцию, а с другой, - повышает фибринолитическую активность крови. Активация этой системы рассматривается как первичный антикоагуляционный механизм. Естестенными антикоагулянтами внешнего пути являются липопротеин-ассоциированный коагуляционный ингибитор (полипептид) - ингибитор внешнего пути свертывания, аполипопротеин А и протеазный ингибитор (антитромбопластины), уменьшающие активность ф.IIа и VIIа, липидный ингибитор - конкурентный ингибитор ф.3 тромбоцитов. Механизмы действия этих антикоагулянтов находятся в стадии изучения. Вторичные антикоагулянты образуются в процессе свертывания крови и фибринолиза. К вторичным антикооагулянтам относятся:
26.1.4. Фибринолитическая система крови Фибрин, образующийся в процессе свертывания крови, подвергается расщеплению - фибринолизу. В системе ферментативного фибринолиза центральным является процесс активации плазминогена с образованием активного плазмина - главного фермента фибринолитической системы (рис.26-5). 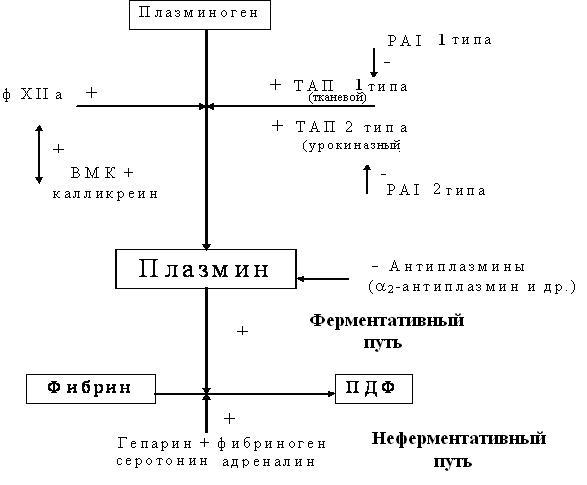 Рис. 26-5. Взаимодействие свертывающей и фибринолитической систем крови Обозначения: ВМК- высокомолекулярный кининоген, ТАП- тканевой активатор плазминогена, РАI - ингибитор активатора плазминогена, (+) - активирующее влияние, (–) - ингибирующее влияние. Примечание: при патологических процессах возможна как первичная, так и вторичная активация фибринолиза . Вещества, вызывающие эту реакцию, называют активаторами плазминогена. Они обнаружены во многих тканях и биологических жидкостях организма. Выделяют 2 типа тканевых активаторов плазминогена (ТАП) - тканевой (ТАП 1) и урокиназный (ТАП 2). Эти активаторы плазминогена представляют собой сериновые протеазы, которые синтезируются, главным образом, в эндотелиальных клетках, а также образуются в процессе микросомального и лизосомального окисления во всех органах, за исключением печени. Наибольшее количество активатора образуется в матке, щитовидной железе, надпочечниках, легких, предстательной железе. ТАП 1 синтезируется в моноцитах, гистиофагах и выделяется в кровь в небольших количествах. В эпителиальных клетках почечных канальцев образуется урокиназа (ТАП 2), которая вызывает активацию циркулирующего плазминогена и определяет 15% внешней фибринолитической активности против 85 %, отмечаемой у ТАП 1. Самостоятельной фибринолитической активностью обладают протеазы лейкоцитов. При стрессе, физической нагрузке, введении некоторых фармакологических препаратов (АДГ, катехоламины, препараты, содержащие никотиновую кислоту) активность активатора плазминогена в циркулирующей крови быстро возрастает. Мощными стимуляторами выделения ТАП 1 являются вазоактивные вещества, особенно, адреналин и гистамин. Высвобождающийся при повреждении тканей ТАП 1 вызывает локальный фибринолиз, фактически не влияющий на суммарную фибринолитическую активность крови (рис.26-5). Фибринолиз является защитным механизмом, препятствующим избыточному отложению фибрина и тем самым сохраняющим нормальные условия для микроциркуляции. Фибринолитическая активность крови зависит не только от содержания плазминогена и его активаторов, но и от ингибиторов фибринолиза. Ингибиторы фибринолиза делят на антиактиваторы и антиплазмины. Антиактиваторы бывают первого (1) и второго (2) типов. Ингибиторы активатора плазминогена 1 типа продуцируются эндотелиоцитами, гепатоцитами и связывают ТАП. Их выработка повышена у больных инфарктом миокарда, при воспалительных процессах. Ингибитор активатора плазминогена 2 типа, образующийся в эндотелиоцитах, моноцитах и гистиофагах (в т.ч. плаценты), угнетает урокиназную активность. Большие количества ингибитора активатора плазминогена второго типа продуцируются клетками злокачественных опухолей. Антиактиваторы тормозят активацию плазминогена, оказывая преимущественно местное действие. В последние годы определенное значение придают ингибитору фибринолиза, активируемого тромбином (TAFI- thrombin activator fibrinolys inhibitor). Он представлен белком, который после активации тромбином приобретает антифибринолитическую активность. Антиплазмины инактивируют плазмин и находятся в плазме в избытке. К ним относятся: 2-макроглобулин, 1-антитрипсин, комплекс АТ III - гепарин и др. Наибольшее значение как физиологический ингибитор плазмина имеет антиплазмин - -2-гликопротеид, образующийся в печени. Антиплазмин в течение 0,1 c необратимо нейтрализует циркулирующий плазмин, а также препятствует связыванию плазмина с фибрином и, таким образом, оказывает дополнительное антифибринолитическое действие. 2-макроглобулин инактивирует плазмин, который взаимодействует с фибрином. Кроме фибринолиза, связанного с действием плазмина (образующегося из плазминогена под влиянием перечисленных выше активаторов) выделяют гепаринзависимый фибринолиз - ф. ХII-зависимый фибринолиз (опосредованный калликреином, комплементом и адреналином). Гепаринзависимый фибринолиз развивается после образования комплексов, в состав которых могут включаться АТ III-гепарин, тромбин, плазминоген, плазмин, фибриноген, катехоламины, серотонин, ф.ХIII, тироксин. Эти комплексы обладают литическим действием на нестабильный фибрин, препятствуют полимеризации фибрин-мономера и стабилизации его ф.ХIII. Считается, что -аминокапроновая кислота тормозит именно гепаринзависимый фибринолиз. Протеазы, высвобождающиеся из гранул активированных лейкоцитов, особенно нейтрофилов, протеазы микроорганизмов и грибов (стрептокиназа, бриназа, охраза), протеазы поджелудочной железы (трипсин, химотрипсин) обладают самостоятельной фибринолитической активностью. Активация фибринолиза отмечается также под действием ФНОα, ИЛ-1 и др. Комплемент-опосредованный фибринолиз возникает при активации фрагмента С8, приводящей к превращению плазминогена в плазмин, а также к активации С3 (компонент С3а участвует в лизисе фибринового сгустка). Значительное повышение фибринолитической активности крови в норме компенсируется нейтрализацией плазмина и усилением элиминации активаторов плазминогена. При патологических процессах возможна первичная и вторичная активация фибринолиза . 26.2. Тромбофилия и ее механизмы Тромбофилия - состояние, характеризующееся предрасположенностью к развитию тромбообразования (тромбозу). Для развития тромбофилии имеют значение повреждения сосудистых стенок, особенно их интимы (повышение тромбогенной активности и снижение тромборезистентности сосудов), тромбоцитарные (повышение функциональной активности тромбоцитов и тромбоцитозы) и плазменные механизмы (увеличение содержания активных коагулянтов в крови (приводящее к гиперкоагуляции); уменьшение антикоагулянтной активности крови и угнетение фибринолиза. 26.2.1. Изменения тромбогенной и тромборезистентной активности сосудистой стенки В клеточных элементах сосудистой стенки образуются такие вещества, как тканевой тромбопластин (ф. III), фибронектин, фактор Виллебранда, тромбоксан А2, фактор активации тромбоцитов (ФАТ) и другие, которые формируют тромбогенный потенциал. Одни из них инициируют адгезию и агрегацию тромбоцитов, другие - тромбиногенез и образование фибрина. В физиологических условиях образование и выделение тромбогенных факторов в сосудистой стенке ограничено, а при ее повреждении или активации эндотелия биологически активными веществами (адреналин, гистамин, серотонин, брадикинин, ИЛ-1, тромбин и др.) значительно увеличивается. В механизме повышения тромбогенности сосудов большое значение имеет интрамуральный тромбоз, развивающийся при микроповреждениях интимы. При этом из тромбоцитов высвобождаются различные факторы, в том числе и фактор роста (ф.4), который вызывает пролиферацию гладкомышечных клеток и фибробластов, миграцию их в интиму, усиление секреции коллагена и других компонентов соединительной ткани, обладающих тромбогенными свойствами. Этот механизм повреждения сосудистой стенки наиболее выражен при сахарном диабете и при атеросклерозе. Эндотелиальные клетки являются одновременно продуцентами и эффекторами ИЛ-1, ИЛ-6, ИЛ-8, ФНОα, рост их продукции отмечается при сепсиcе, опухолях, воспалении и служат неспецифическим ответом на повреждение эндотелия. Это приводит к росту адгезивных свойств эндотелиальных клеток, угнетению активности тромбомодулина на эндотелии, а также к повышению продукции ф. III, что приводит к формированию тромбозов. В последние годы достаточно широко обсуждается значение синдрома антифосфолипидных антител в патогенезе тромбозов и гиперкоагуляции. Впервые они были обнаружены у больных системной красной волчанкой, у которых отмечались тромбозы не только вен, но и артерий. В настоящее время определенное значение имеют антифосфолипидные АТ в патогенезе ИБС. Определенную роль в повреждении эндотелия может сыграть и табакокурение. В сложном механизме повреждения эндотелия сосудов важное значение придают уменьшению продукции моноокиси азота, росту чувствительности и росту выделения эндотелиоцитами вазоконстрикторов, а также снижению ангиопротективной роли ЛПВП из-за уменьшения их синтеза в печени. В последнее время в повреждении сосудистой стенки большое значение также уделяют нарушению обмена аминокислоты гомоцистеина (гипергомоцистеинемии), которое часто (в 10-15% случаев) наблюдается в европейской популяции. При этом инициируется оксидативный стресс и связанное с ним повреждение эндотелия, а также стимулируются процессы пролиферации гладкомышечных клеток сосудов, что способствует развитию атеросклероза. Риск повреждения сосудистой стенки при гипергомоцистеинемии сопоставим с действием таких факторов как гиперхолестеринемия, артериальная гипертензия и табакокурение . Наряду с тромбогенными свойствами сосуды обладают и антитромбогенными свойствами или тромборезистентностью (рис. 26-6). 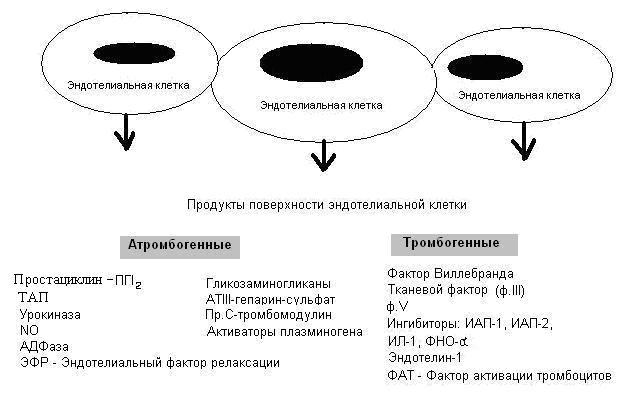 Рис. 26-6. Эндотелиальные клетки и их атромбогенные и тромбогенные продукты. В эндотелии и в меньшей степени в других клетках сосудистой стенки образуется ряд факторов, которые ингибируют свертывание крови, активируют фибринолиз, тормозят адгезию и агрегацию тромбоцитов. К факторам, обладающим антикоагулянтной активностью и образующимся в основном в эндотелии, относятся протеины С и S, тромбомодулин, гепаринсульфаты и другие. Именно гликозаминогликаны и составляют выраженный отрицательный заряд эндотелия сосудистой стенки, препятствующий адгезии форменных элементов крови на последней. Активация фибринолиза происходит при участии сосудистого активатора плазминогена и протеина С. Торможение адгезии и агрегации тромбоцитов происходит под влиянием простациклина, протеогликанов, фактора релаксации эндотелия, NО. Повышение тромбогенной активности сосудистой стенки и снижение тромборезистентности создают условия для тромбоза даже при незначительном повреждении сосуда. 26.2.2. Повышение функциональной активности тромбоцитов и тромбоцитозы Повышенная наклонность тромбоцитов к адгезии и агрегации наблюдается при атеросклерозе, сахарном диабете, гипертонической болезни и других заболеваниях. Усиление агрегации тромбоцитов может быть первичной, т.е. связанным, например, с гиперпродукцией тромбоксана А2 (которая отмечается при инсулинзависимом сахарном диабете, позднем гестозе беременных, злокачественных опухолях и др.). Гипергликемия, гиперлипидемия (особенно, за счет ЛПНП и ЛПОНП) способствуют повышению агрегационной активности тромбоцитов и их чувствительности к индукторам агрегации. При инсулинзависимом сахарном диабете это обусловлено наличием гликозилированных рецепторов тромбоцитов, что обеспечивает их большую реактивность, в том числе и активную адгезию к коллагену сосудистой стенки. Увеличение содержания в крови адреналина, -липопротеидов, свободных жирных кислот, ф.W сопровождается значительным повышением агрегационной активности тромбоцитов. Дефицит простациклина, как основного антиагрегантного вещества, также характеризуется гиперагрегацией тромбоцитов. Тромбоцитозы, особенно, рост числа тромбоцитов выше 600٠109 обычно вызывают тромбофилию. Тромбоцитозы отмечаются при некоторых миелопролиферативных заболеваниях (болезнь Вакеза, миелофиброз), после спленэктомии, при дефиците железа и т.д.. Для тромбогенных ситуаций, как правило, характерно снижение порога чувствительности тромбоцитов к индукторам агрегации, усиление спонтанной агрегации тромбоцитов в кровотоке, уменьшение продолжительности жизни циркулирующих тромбоцитов. В плазме повышается уровень внутритромбоцитарных факторов (-тромбоглобулин, антигепариновый фактор и др.), что указывает на активацию "реакции высвобождения". Существенное значение имеет использование лекарственных ингибиторов рецепторов тромбоцитов к фибриногену IIb/IIIa в ситуациях, когда тромбозы артериол приводят к инфаркту миокарда и другим критическим состояниям. 26.2.3. Гиперкоагуляция и ее механизмы В некоторых случаях, например после острой кровопотери, повышение свертываемости крови является компенсаторной реакцией. Длительная и выраженная гиперкоагуляция создает условия для активации тромбообразования (тромбофилии). Повышение свертываемости крови (гиперкоагуляция) и тромбофилия наблюдаются при многих заболеваниях и патологических процессах. К их развитию могут привести следующие механизмы. 26.2.3.1. Увеличение содержания прокоагулянтов в крови Повышение активности плазменных прокоагулянтов и коагуляционного потенциала само по себе не приводит к тромбообразованию, но при повреждении сосудистой стенки способствует ускорению и распространению тромбоза. В механизме гиперкоагуляции в условиях патологии большое значение имеет поступление в кровь тканевого тромбопластина из сосудистой стенки. Это ведет к образованию протромбиназы и тромбина в количествах, превосходящих антитромбиновую активность крови, в результате повышается коагуляционный потенциал. В мобилизации активаторов свертывания крови из сосудистой стенки при стрессе большое значение имеет адреналин. Активация симпато-адреналовой системы стимулирует синтез фибриногена, а повышение продукции глюкокортикоидов - протромбина, фибриногена, акцелератора-глобулина. Повышение активности прокоагулянтов может быть обусловлено непосредственным воздействием на них некоторых компонентов плазмы. Гиперлипидемия создает условия для спонтанной активации ф.ХII и ускорения образования протромбиназы. Гиперлипидемия и тромбопластинемия отмечаются после интенсивной эмоциональной и физическоой нагрузки, поэтому у спортсменов-профессионалов возможны острые тромбозы различных (в т.ч. коронарных) артерий при уменьшении тромборезистентных свойств сосудистой стенки. У больных с клиническими проявлениями атеросклероза и гипертонической болезни отмечается значительное увеличение в крови фибриногена, протромбина, ф.VIII, ф.XII и др. Массивное поступление в кровь тканевого тромбопластина (в результате обширного повреждения тканей, гемолиза), как и внутрисосудистая активация ф.XII при септицемии могут привести к внутрисосудистому свертыванию крови и нарушению гемодинамики. Выраженное тромбообразование вследствие повреждения микрососудов, отмечаемое в зоне имплантата, также может приводить к остеодеструкции вследствие некроза. У беременных женщин с конца второго и начала третьего триместра отмечается прогрессирующий рост в плазме факторов свертывания I, II, VII, VIII, IX, XI, XII. К концу физиологически протекающей беременности их уровень повышается в среднем до 150-300%, а концентрация фибриногена может даже достигать 4-5 г/л. Следует отметить, что для дисфибриногенемии, наследуемой аутосомно-рецессивным путем, характерен аномальный синтез молекул фибриногена. 26.2.3.2. Снижение антикоагулянтной активности крови Одной из причин тромбофилии является дефицит естественных антикоагулянтов, прежде всего, АТ III. Нарушение метаболизма и снижение количества АТ III может быть врожденным и приобретенным. Врожденный дефицит АТ III наследуется по аутосомно-доминантному типу и проявляется в уменьшении как выработки АТ III, так и его сродства к гепарину и тромбину. Приобретенная недостаточность АТ III развивается у больных с печеночной недостаточностью и преимущественным снижением синтетической функции гепатоцитов, в том числе обусловленной дефицитом витамина А. Вариантом приобретенной недоостаточности АТ III является истощение его депо у больных с хронической почечной недостаточностью, с нефротическим синдромом, острым венозным тромбозом, ДВС-синдромом, на фоне интенсивной гепаринотерапии. Возможно связывание АТ III антителами. При атеросклерозе, сахарном диабете, поздних стадиях гипертонической болезни уменьшается содержание гепарина в крови. Это связано с истощением эндогенных ресурсов гепарина в результате постоянного расхода его в качестве кофермента липопротеиновой липазы. Одной из причин гепарино-резистентности также может быть нарушение взаимодействия АТ III с гепарином, отмечаемое у больных с системной красной волчанкой, синдромом Шенлейна-Геноха. Врожденные дефициты ПрС и ПрS наследуются аутосомно-доминантным путем, частота встречаемости в популяции составляет 1:300. Уменьшение содержания ПрS в крови описано при нормальной беременности, приеме гормональных контрацептивных препаратов, тромбозе спланхнических вен и является фактором риска тромбоза и гиперкоагуляции. |