_Спирты. Спирты
 Скачать 1.03 Mb. Скачать 1.03 Mb.
|

|
13.4. Методы синтеза одноатомных спиртов в лаборатории
 Во избежание побочных процессов первоначально из хлоралканов предпочтительно синтезировать сложные эфиры, которые затем омыляют до спиртов. 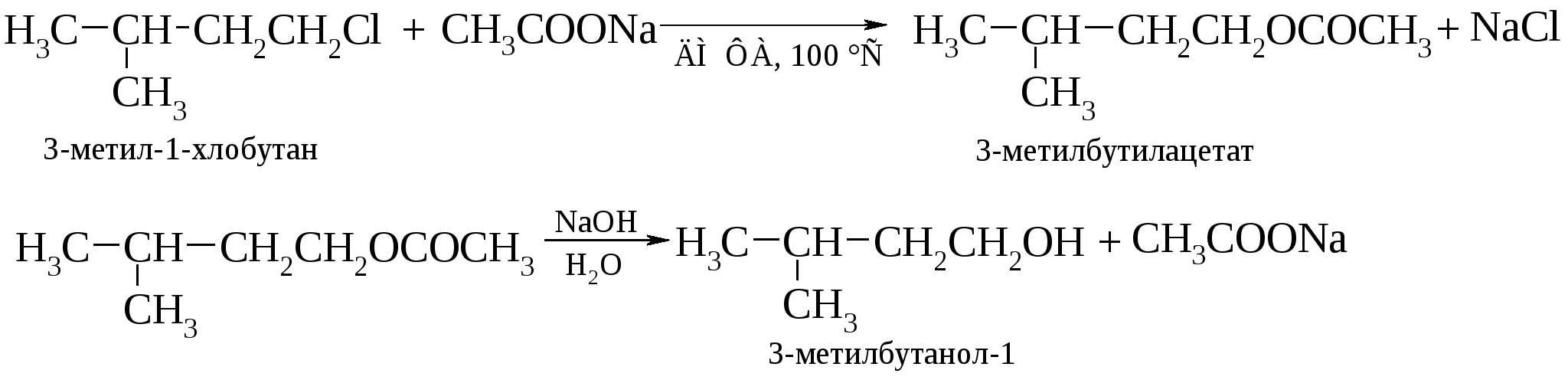 Для лучшей гомогенизации реакционной смеси в нее добавляют некоторое количество смешивающегося с водой растворителя, например, диоксана.
Реакцию проводят в тетрагидрофуране. Диборан получают реакцией между двумя коммерческими реагентами: боргидридом натрия и фторидом бора, часто insitu (в реакционной смечи в присутствии алкена) или восстанавливают хлорид бора(III) водородом. Алкилбораны не выделяют, а обрабатывают их в том же реакционном сосуде щелочным раствором пероксида водорода. Как видно из приведенных реакций, они протекают против классического правила Марковникова и без перегруппировок.  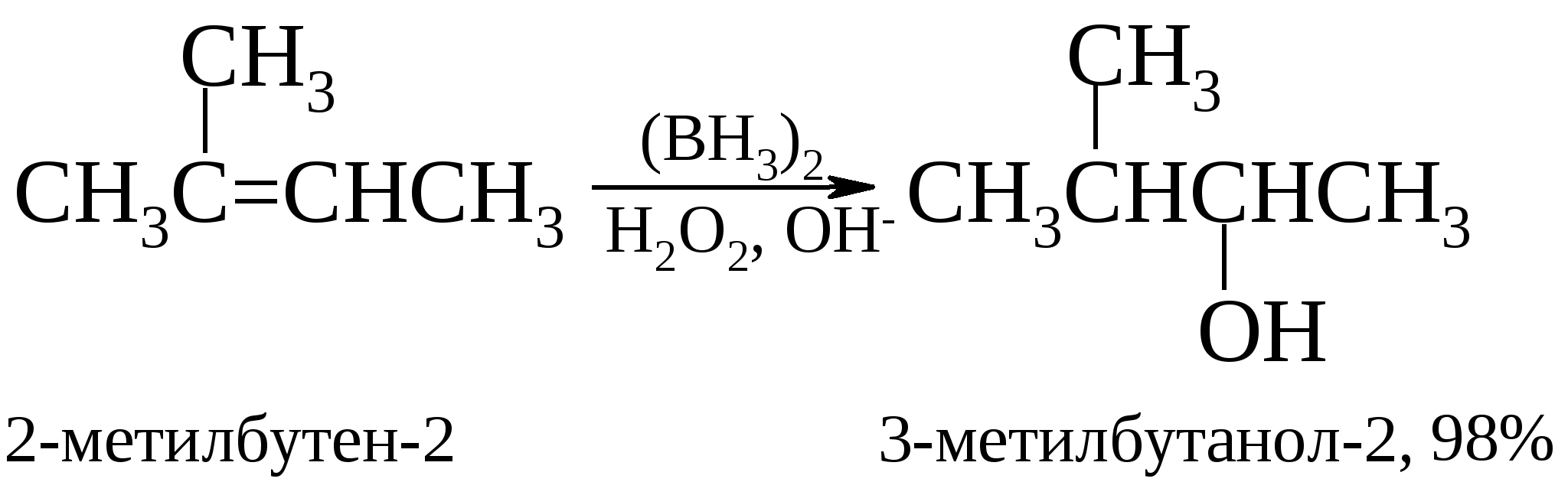 Следует отметить, что в реакции участвует не диборан, а его мономер, образующийся в растворе: Наряду с дибораном в органическом синтезе применяют комплекс борана в тетрагидрофуране.
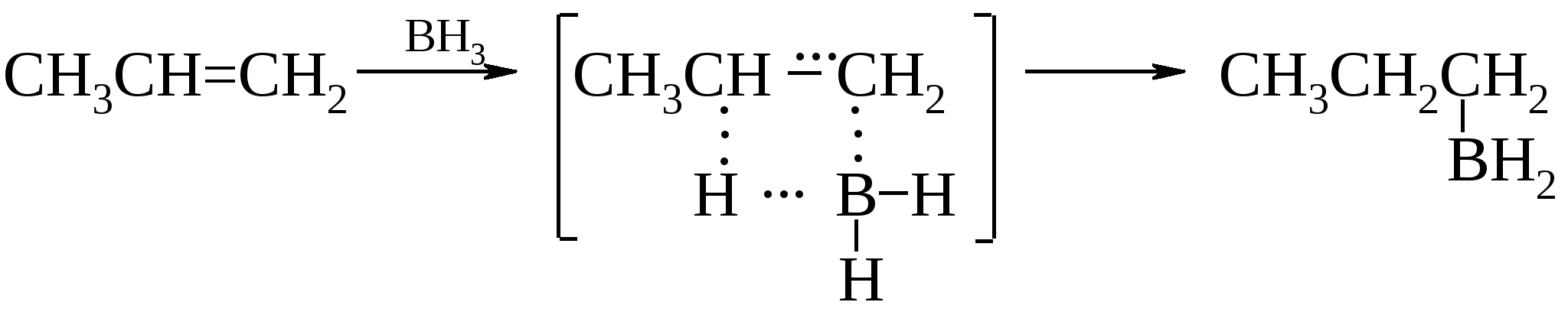 По-видимому, реакция гидроборирования алкенов начинается с электрофильной атаки атома бора. В образовавшемся -комплексе на атоме бора увеличивается отрицательный заряд с тенденцией образования вторичного карбкатиона. Однако последний не образуется, потому что приобретающий отрицательный заряд атом бора легко теряет атом водорода в виде гидрид-иона с синхронным образованием продукта цис-присоединения. Реакция окисления алкилборанов протекает следующим образом. На первой стадии гидропероксид-анион атакует электронодефицитный атом бора. 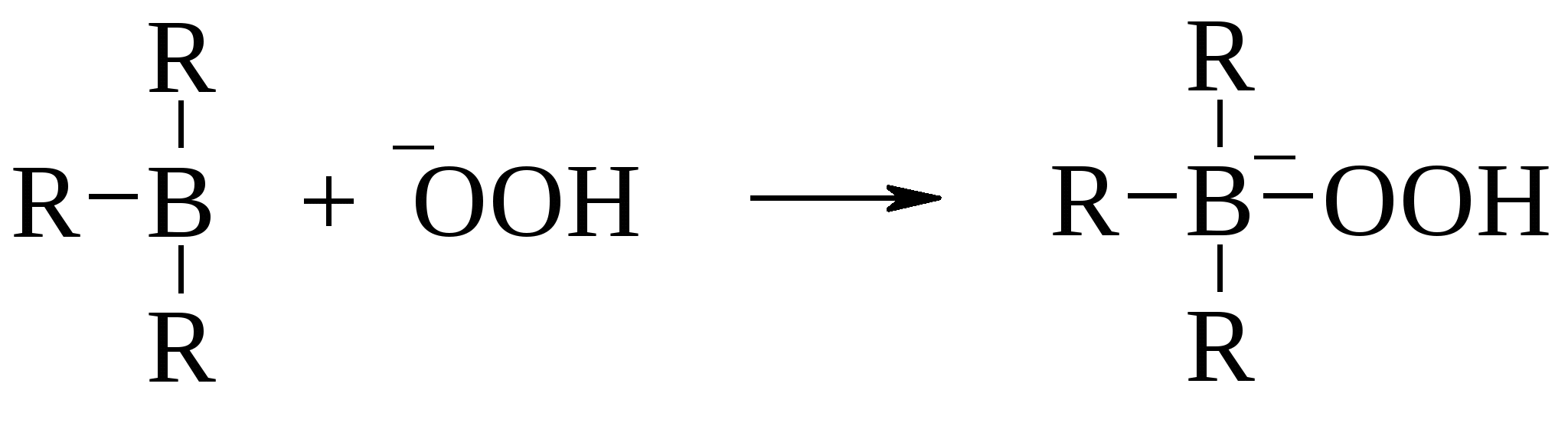 Образовавшийся интермедиат перегруппировывается благодаря миграции алкильной группы со своими электронами к атому кислорода по схеме, сходной с перегруппировкой карбкатионов. 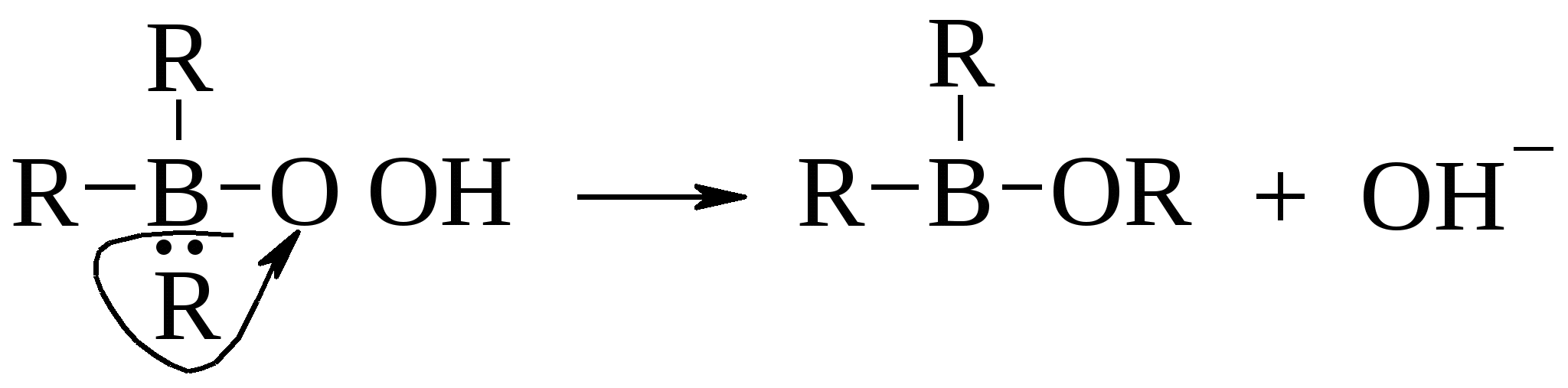 Взаимодействие с гидропероксидом в щелочной среде протекает быстро и с выделением тепла. Образовавшийся эфир борной кислоты легко разлагается в условиях реакции с высвобождением спирта. Чтобы предотвратить дальнейшее окисление продуктов реакции до альдегидов и кислот, процесс проводят в атмосфере азота в присутствии борной кислоты (А. Башкиров), которая образует со спиртами стойкие к окислению эфиры борной кислоты В(ОR)3. Последние затем легко гидролизуются щелочами. Таким путем в промышленности, в частности, получают цетиловый спирт С16Н33ОН. Реакция гидроборирования проста и удобна, выходы очень высоки, и ее можно использовать для синтеза соединений, которые трудно получить из алкенов каким-либо другим способом. Для ациклических, моно- и дизамещенных алкенов гидроборирование–окисление дает уникальную возможность синтеза первичных спиртов с суммарным выходом 80-95%.
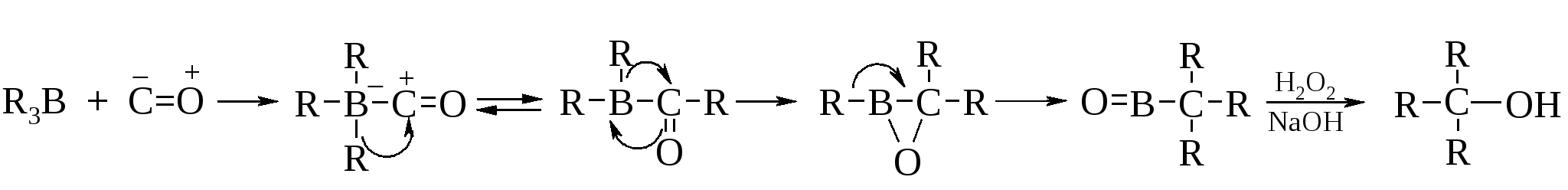 Этим методом в зависимости от условий проведения реакции можно получить с высоким выходом первичные, вторичные и третичные спирты.
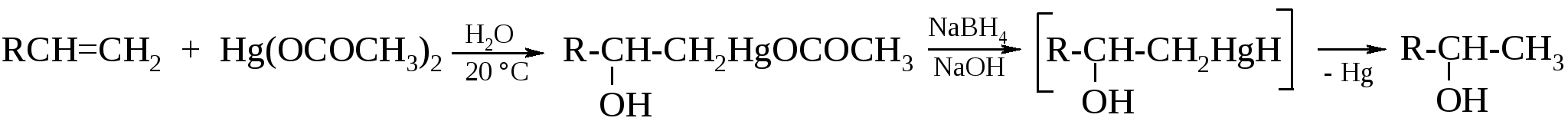 Механизм этой реакции может быть представлен следующим образом. Первоначально происходит диссоциация ацетата ртути(II) с образованием иона СН3СООНg+. Ацетоксимеркурат-катион реагирует с двойной связью С=С алкена подобно протону. Далее карбокатион взаимодействует с водой, образуя алкилртутную соль. Механизм этой реакции может быть представлен следующим образом. Первоначально происходит диссоциация ацетата ртути(II) с образованием иона СН3СООНg+. Ацетоксимеркурат-катион реагирует с двойной связью С=С алкена подобно протону. Далее карбокатион взаимодействует с водой, образуя алкилртутную соль.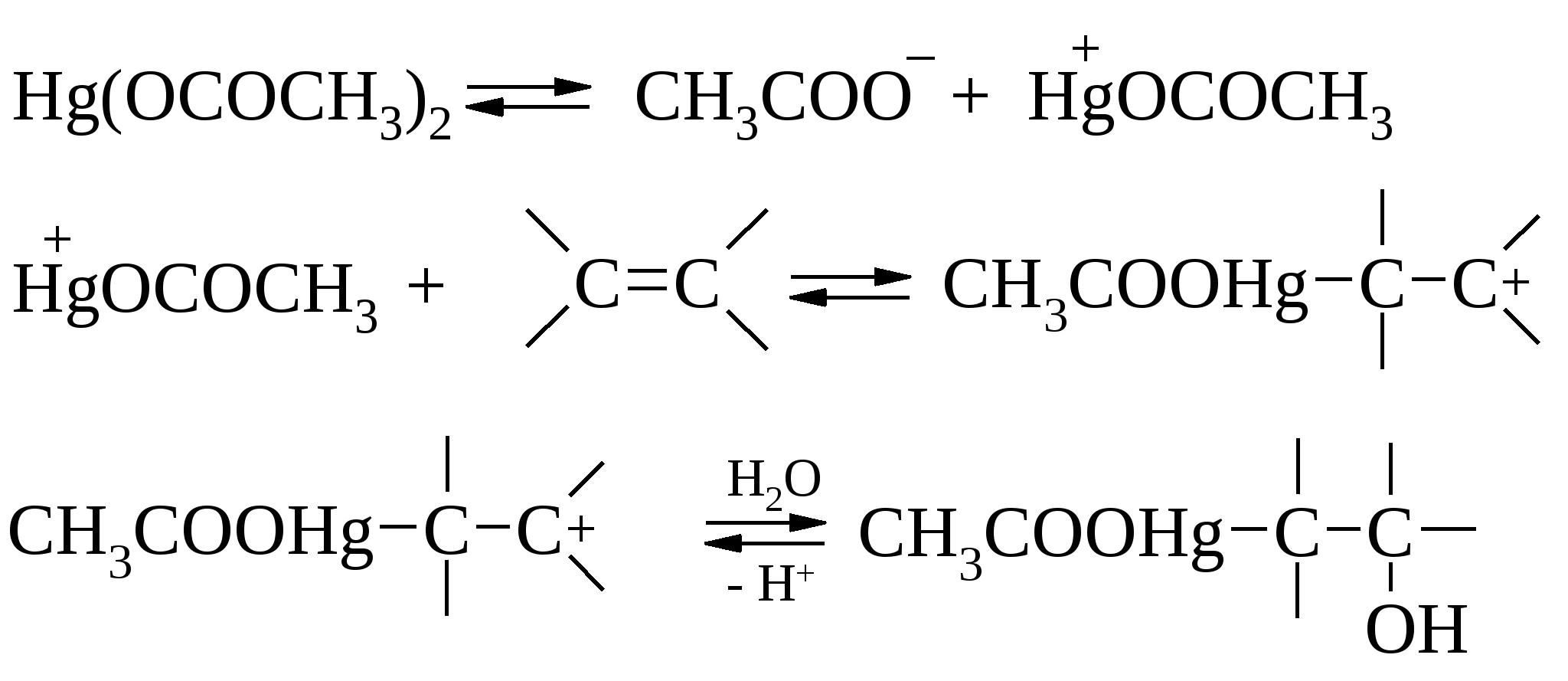 Демеркурирование образующихся меркурированных спиртов протекает количественно при обработке их боргидридом натрия. 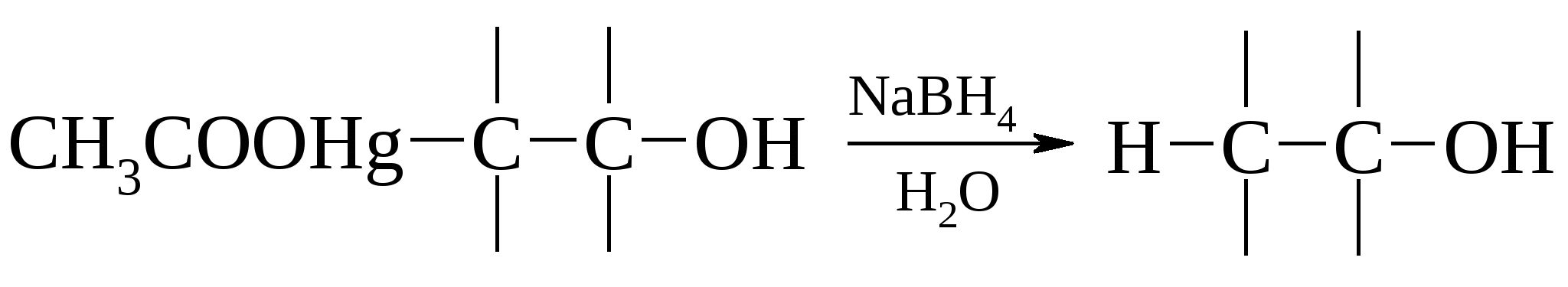 Например: 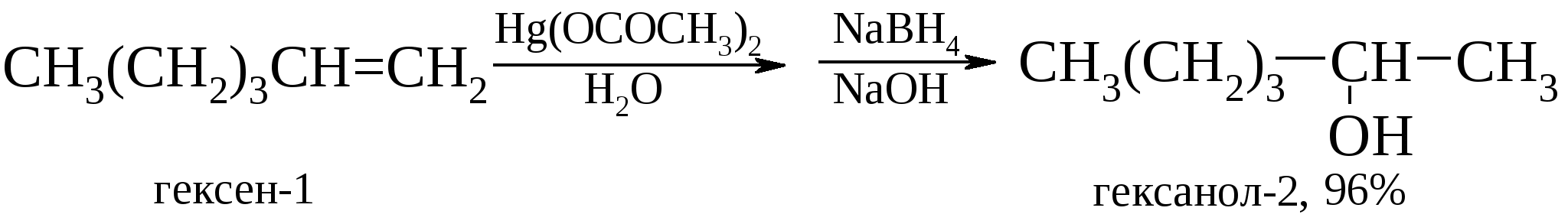 Замена воды на спирт или карбоновую кислоту приводит к получению простых или сложных эфиров. В лаборатории этот метод полностью вытеснил реакцию гидратации алкенов.
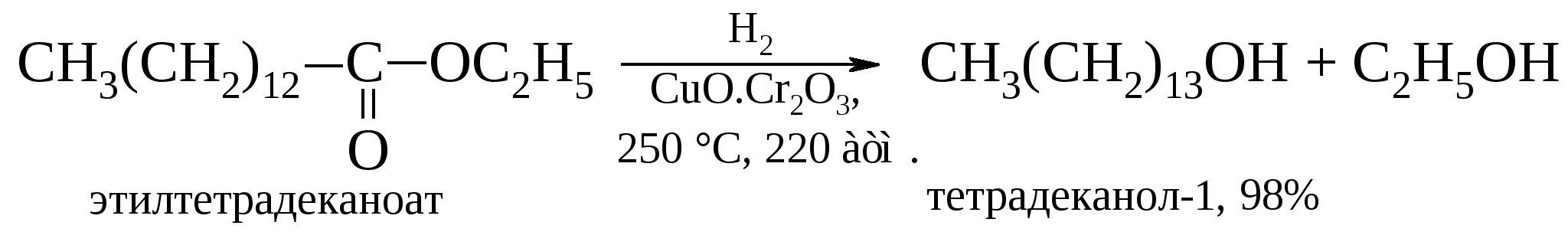
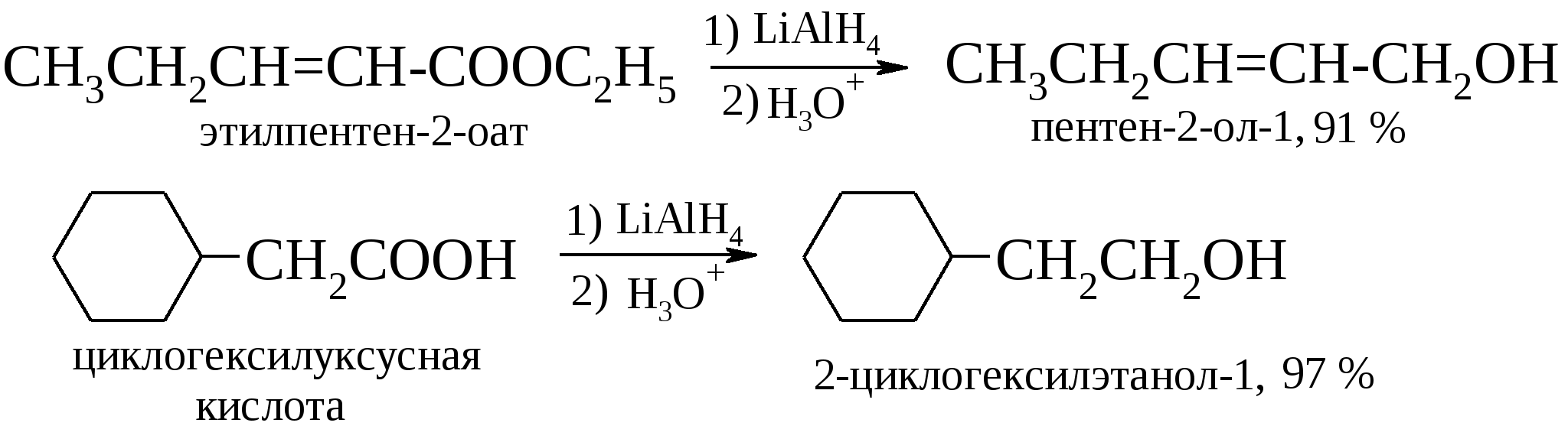
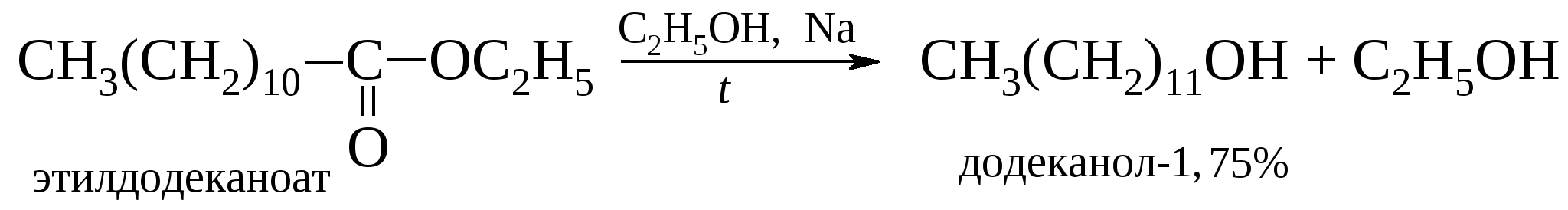
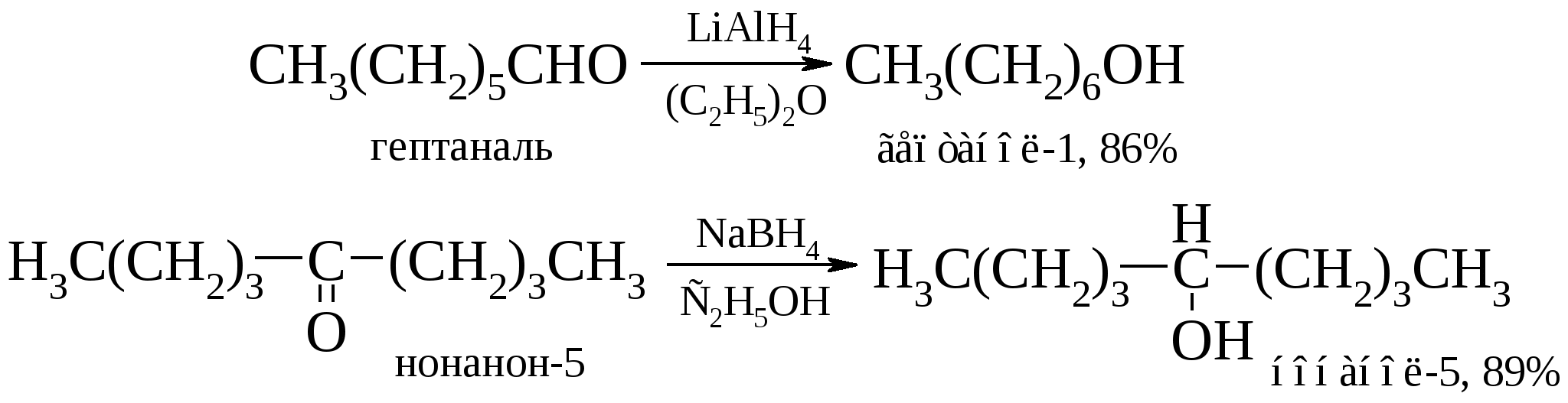 Следует отметить, что боргидрид натрия в отличие алюмогидрида лития не восстанавливает карбоксильную и сложноэфирную группы, что позволяет восстанавливать карбонильную группу в их присутствии. 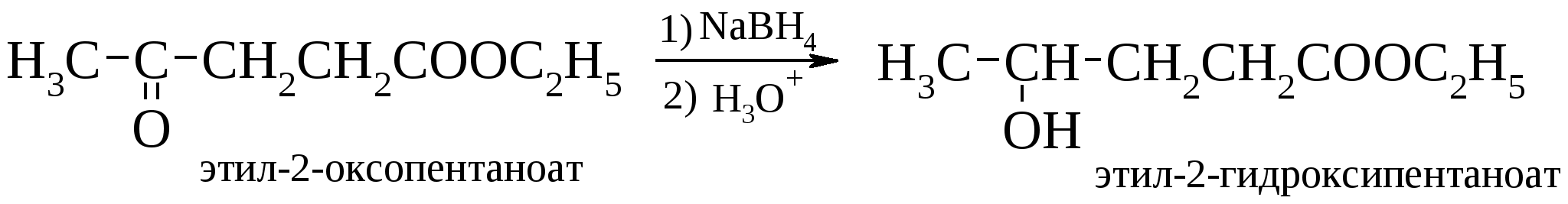 Алкил- и арилзамещенные боргидриды наряду с селективностью восстановления обеспечивают и стереоселективность.
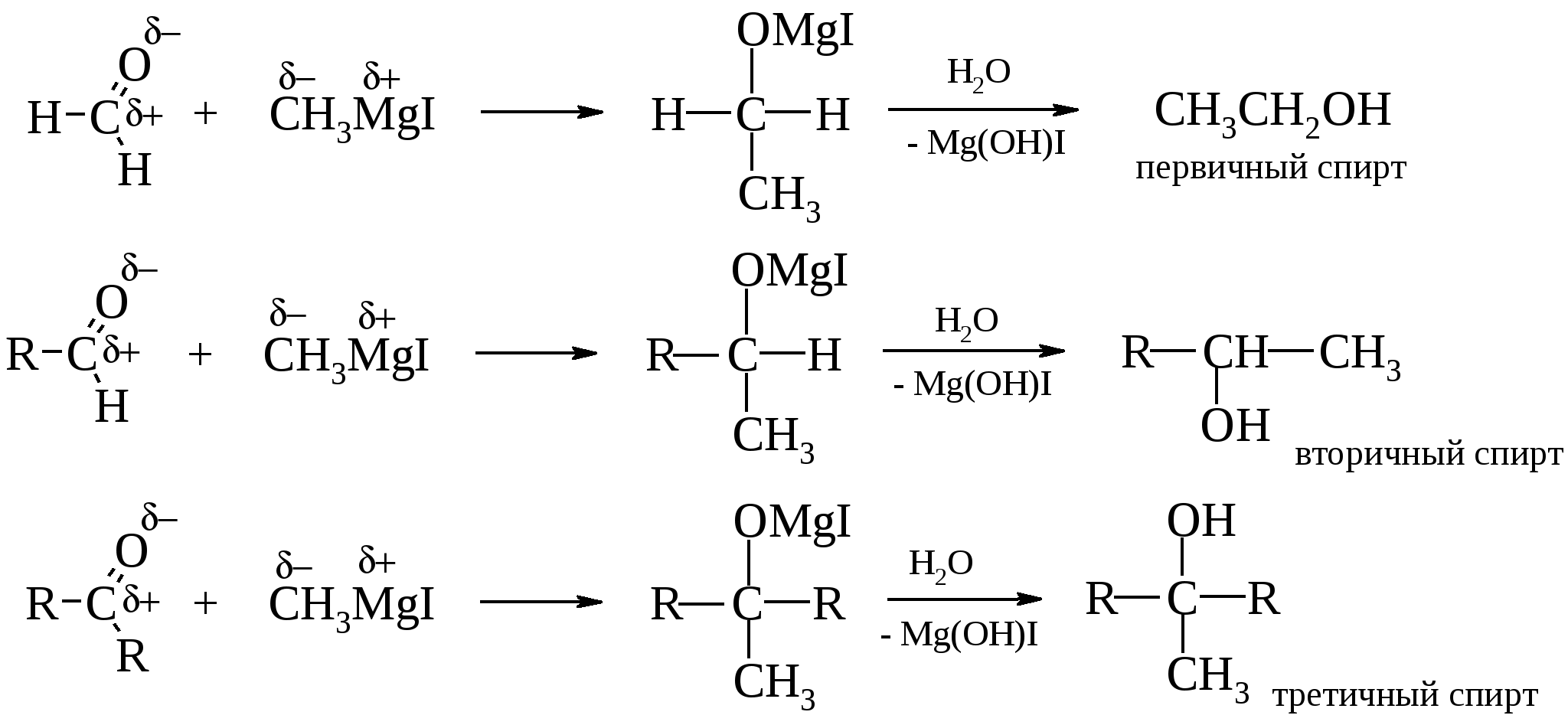 При взаимодействии реактива Гриньяра со сложными эфирами получают третичные спирты, за исключением эфиров муравьиной кислоты, дающих вторичные спирты. 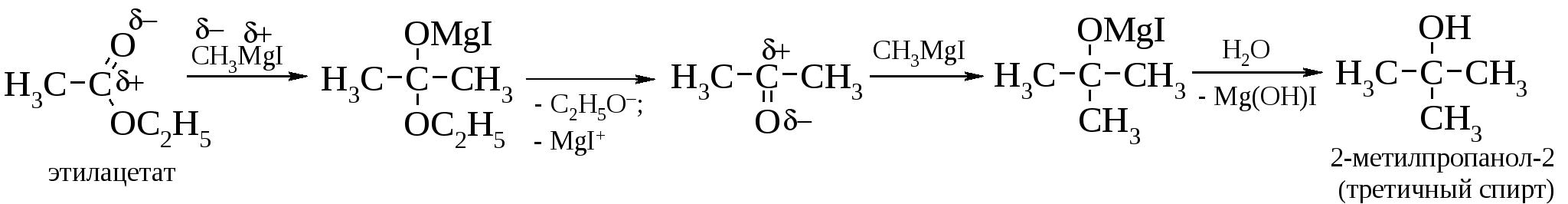 Образовавшийся кетон более активен, чем сложный эфир, и поэтому он в первую очередь реагирует с реактивом Гриньяра. Образовавшийся кетон более активен, чем сложный эфир, и поэтому он в первую очередь реагирует с реактивом Гриньяра.
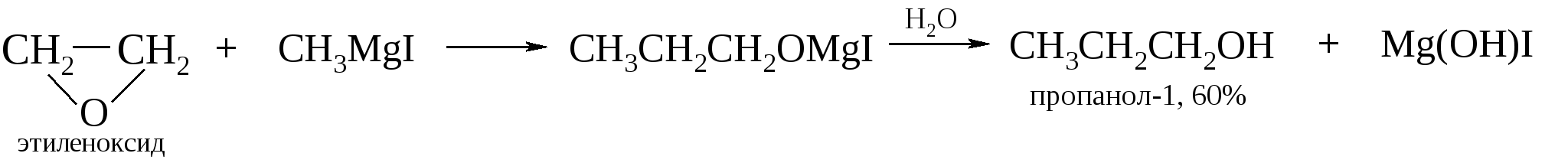
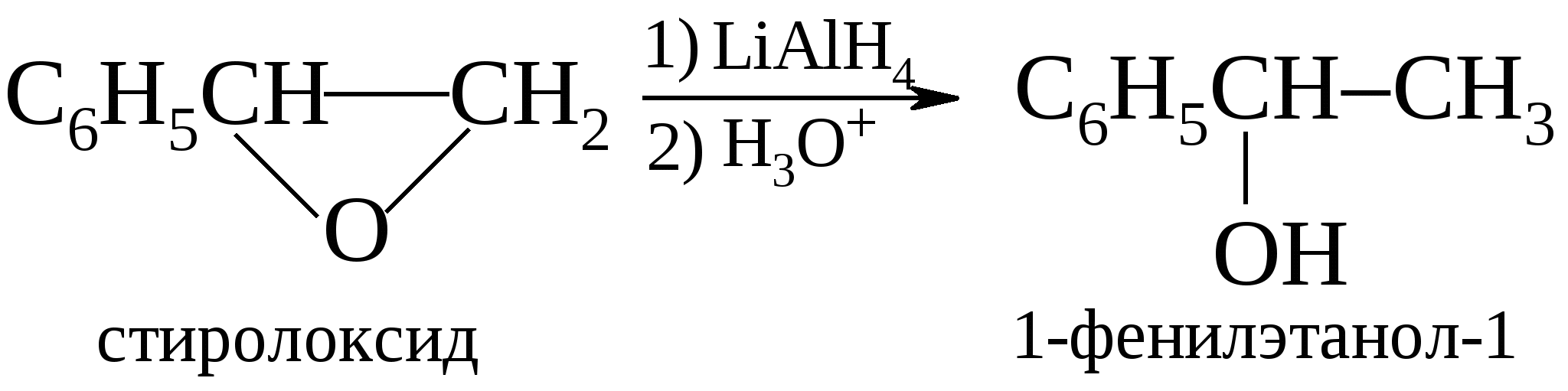 Ввиду того, что -оксиды обычно получают из олефинов, то такой двухстадийный процесс можно рассматривать как альтернативу реакции гидратации алкенов. В отличие от последней реакции восстановление эпоксидов протекает регио- и стереоспецифично. В системах, для которых невозможно свободное вращение вокруг -связей гидроксильная группа и атом водорода имеют анти-конфигурацию, отсюда и название этого процесса – анти-гидратация. 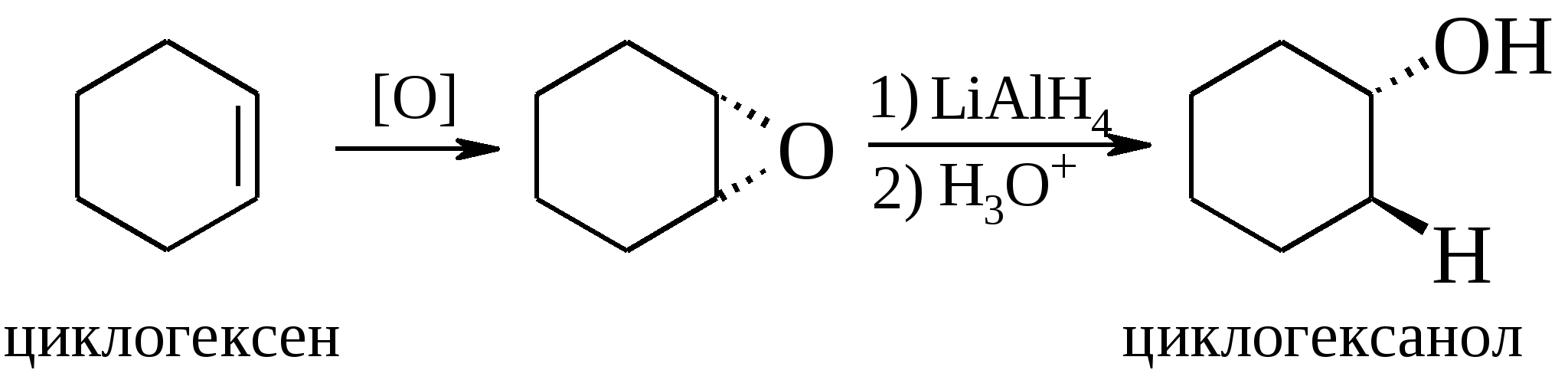
CnH2n+1NH2 + HONO → CnH2n+1OH + N2↑ + H2O Реакция не имеет реальной синтетической значимости, т. к. сопровождается образованием большого количества побочных продуктов.
Замещение атома галогена у вторичных асимметрических атомов углерода на гидроксил сопровождается полным обращением конфигурации. 13.5. Химические свойства одноатомных спиртов Реакции спиртов можно разделить на два типа: протекающие с разрывом связи С–ОН и СО–Н, в силу того, что спирты проявляют кислотно-основные свойства. 13.5.1. Разрыв связи С–ОН
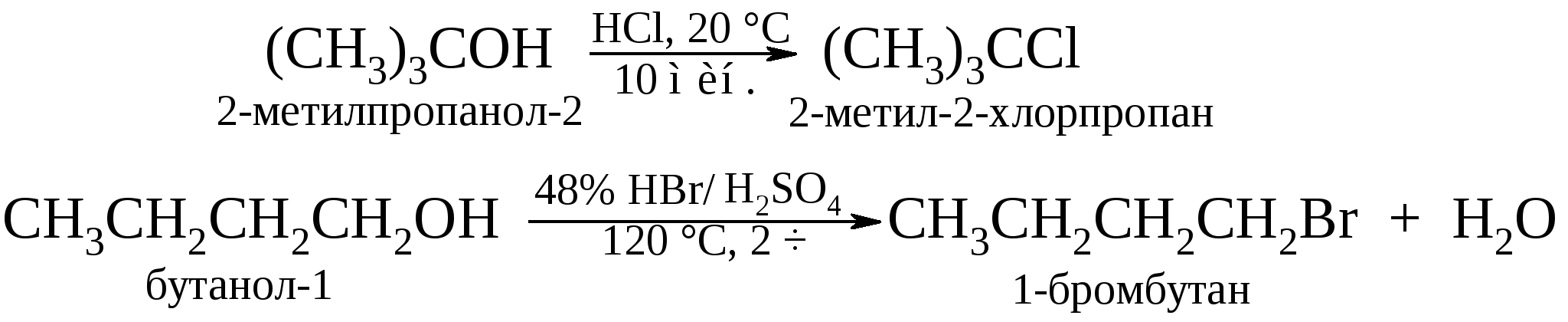 Иногда галогеноводородные кислоты получают в реакционной смеси из их натриевых и калиевых солей действием концентрированной серной кислоты.
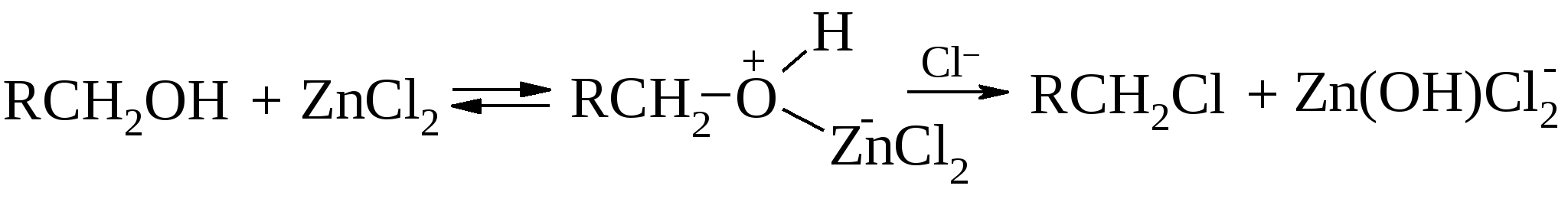 Так, т. е. по механизму SN2, реагируют метанол и большинство пространственно незатрудненных первичных спиртов. Протонирование спиртов превращает гидроксильную группу в хорошо уходящую группу. В реакциях SN2 реакционная способность первичных спиртов R – CH2OH ниже, чем для самого метанола. Это связано с увеличением пространственных затруднений для атаки протонированного спирта галогенид-ионом.
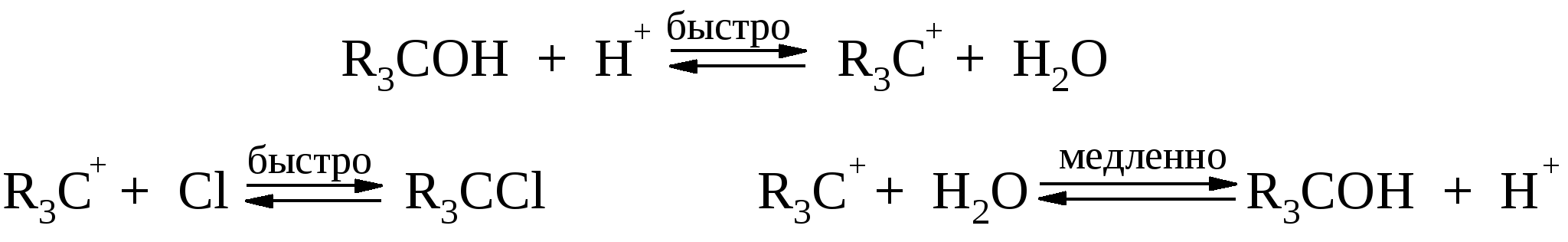
 Последняя стадия осложняется побочной реакцией Е1 – отщеплением протона с образованием алкена. 
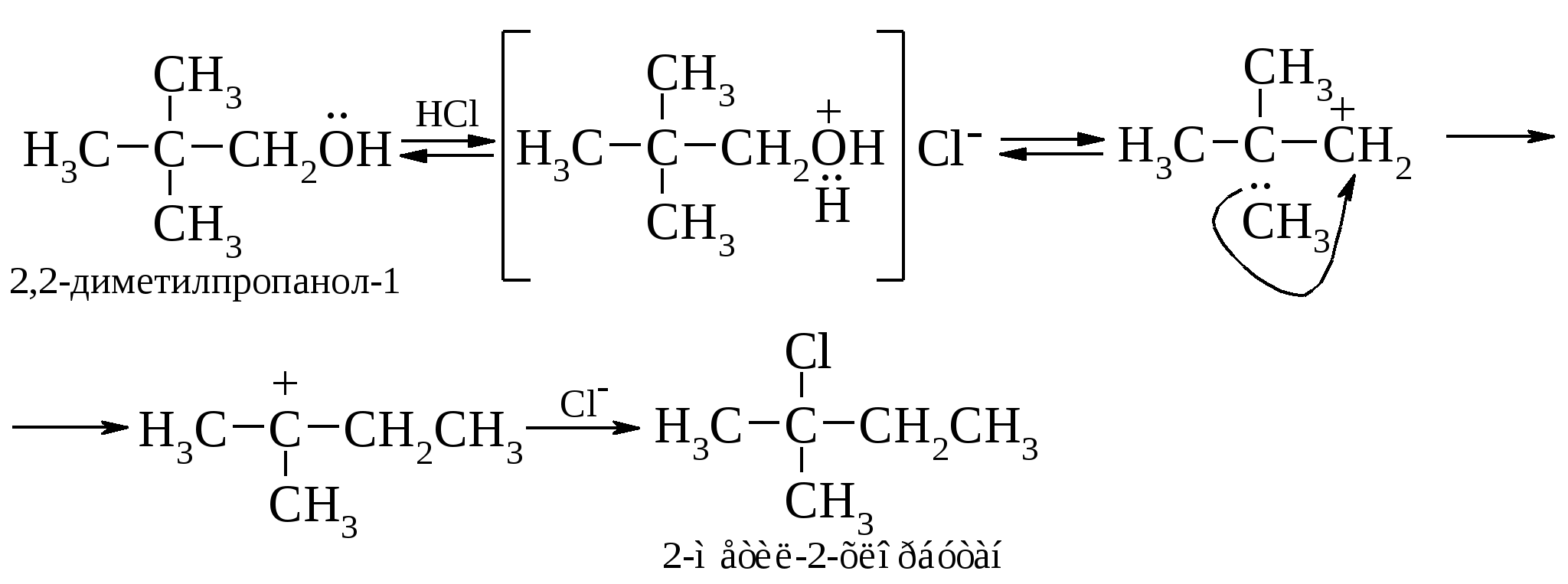 Вторичные спирты могут реагировать как по механизму SN1, так и SN2. Он определяется концентрацией спирта, кислоты, температурой проведения реакции и природой растворителя.
R-OH + PCl5 → R-Cl + POCl3 + HCl 3 R-OH + PBr3 → 3 R-Br + H3PO3 6 CH3OH + 2 P + 3 I2 → 6 CH3I + H3PO3 (P + 3 I2 → 2PI3)
Если бромоводород не нейтрализовать, то промежуточное соединение триалкилфосфит легко протонизируется и алкильные группы превращаются в галогеналканы.

RОН + SOCl 2 → ROSOCl + HCl Далее реакция может протекать по двум механизмам. В нуклеофильных растворителях, таких как диоксан, растворитель принимает участие в реакции и в конечном продукте сохраняется исходная конфигурация. 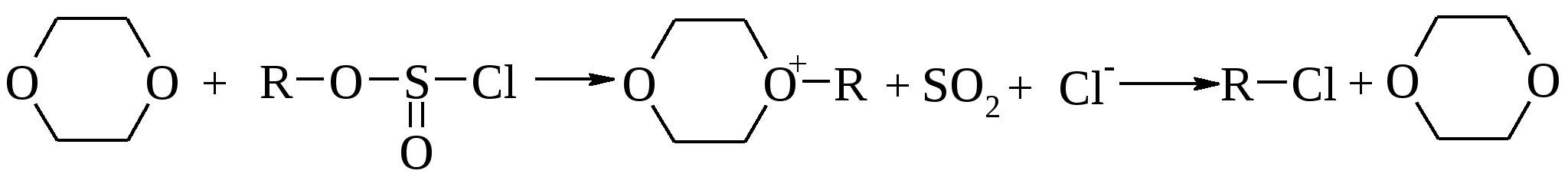 В том случае, когда растворитель не принимает участия в реакции, атака хлорид-аниона молекулы хлорсульфитного эфира протекает с тыла с обращением конфигурации продукта реакции.
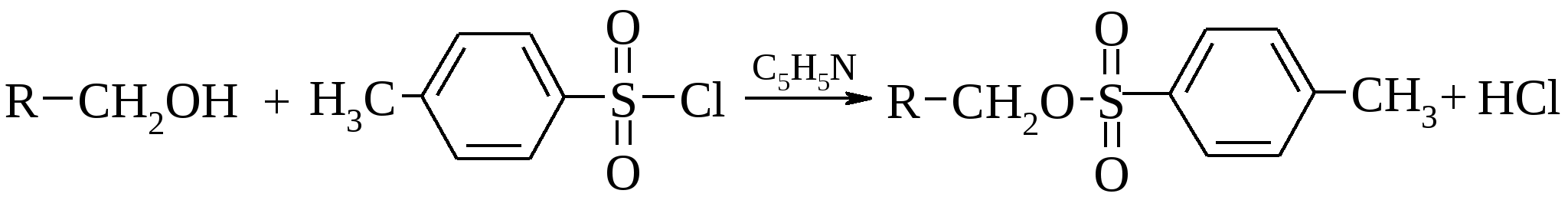 Поскольку п-толуолсульфат-ион является очень легко уходящей группой, то она может быть легко замещена без перегруппировок в реакциях с нуклеофилами, в том числе и с галогенид-ионами.
 Как уже упоминалось ранее, легче всего дегидратируются третичные спирты, потом вторичные и, наконец, первичные. Процесс дегидратации спиртов подчиняется правилу Зайцева, согласно которому атом водорода отщепляется от наименее гидрогенизированного атома углерода, что находится в -положении к ОH-группе, например: 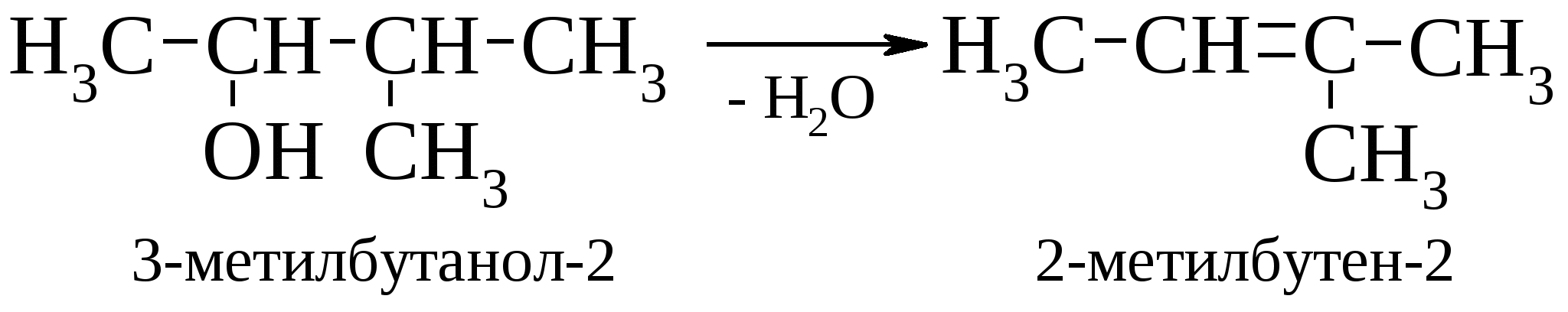 . .Дегидратация спиртов протекает в два этапа. Сначала происходит протонирование ОН-группы, а затем элиминирование молекулы воды по механизму Е2, если речь идет о первичных спиртах, или по Е1 – механизму, если спирты третичные. Вторичные спирты в зависимости от условий реакции могут дегидратироваться по Е2 или Е1 механизму.
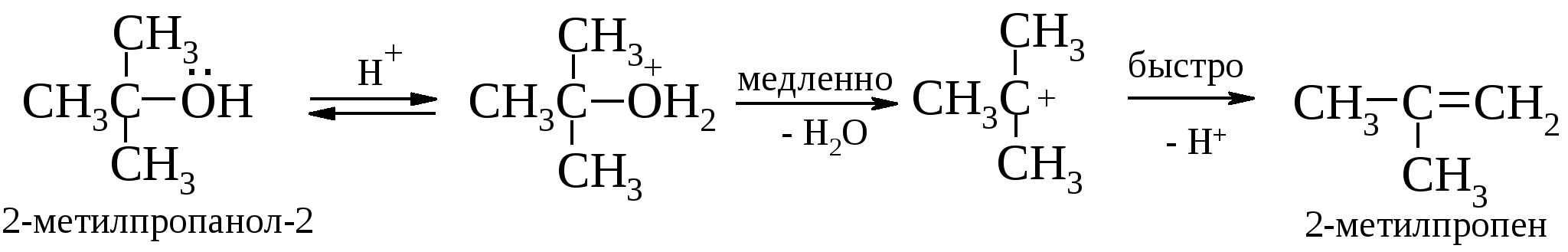 Третичные спирты дегидратируются настолько легко, что возможна избирательная дегидратация диола, содержащего первичную и третичную гидроксильные группы. 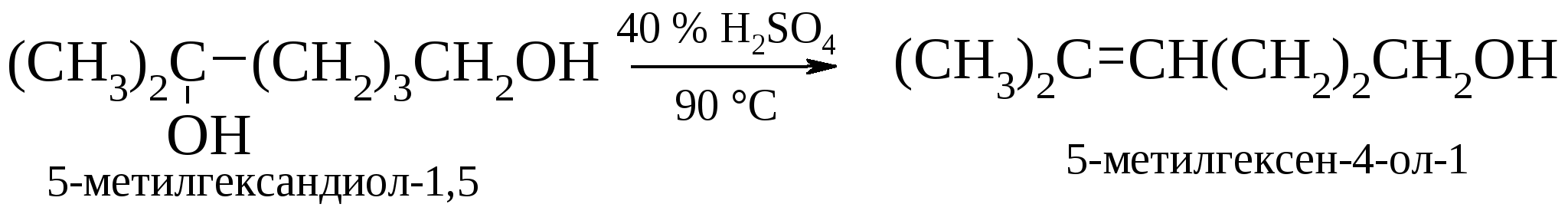 Дегидратацию третичных спиртов можно проводить уже в 20-50%-ной серной кислоте при 85-100 ºС. Вторичные спирты подвергаются дегидратации в более жестких условиях: 85%-ная фосфорная кислота, нагревание до 160 ºС или 60-70%-ная серная кислота при температуре 90-100 ºС.
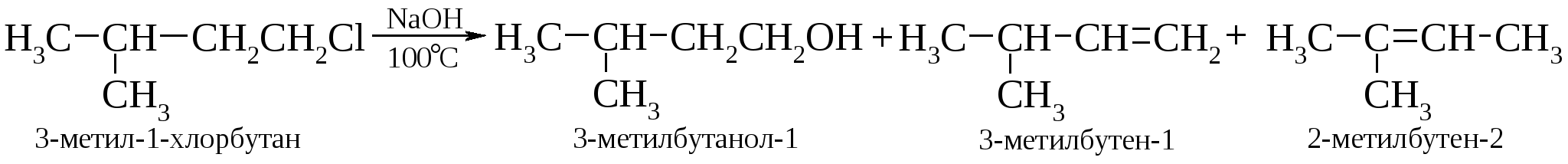
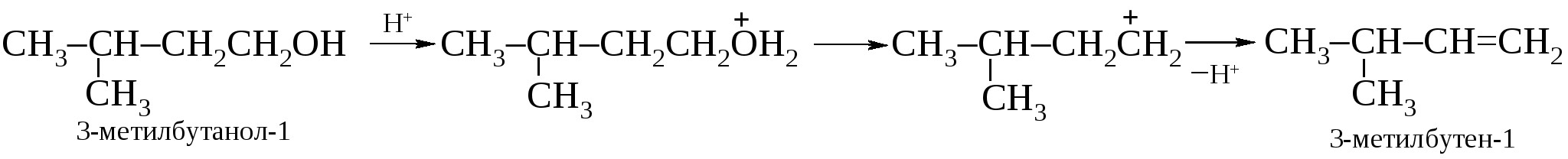 Образовавшийся сначала первичный карбокатион – наименее стабильный, поэтому он в результате 1,2-гидридного сдвига переходит в более стабильный вторичный карбокатион.  В свою очередь, вторичный карбкатион легко превращается в третичный как наиболее стабильный.  Больше всего в продуктах реакции будет 2-метилбутена-2 как наиболее разветвленного алкена. Следует отметить, что изоамиловый спирт относится к первичным спиртам, тем не менее его дегидратация протекает по механизму Е1, что объясняется невозможностью реализации механизма Е2 из-за стерических затруднений.
Для них реализуется механизм отщепления Е2. В реакцию вступает не сам спирт, а алкилсульфат, а роль нуклеофила играет гидросульфат-анион или вода.  Интересно отметить, что при проведении реакции при низкой температуре процесс можно остановить на стадии алкилсульфата. 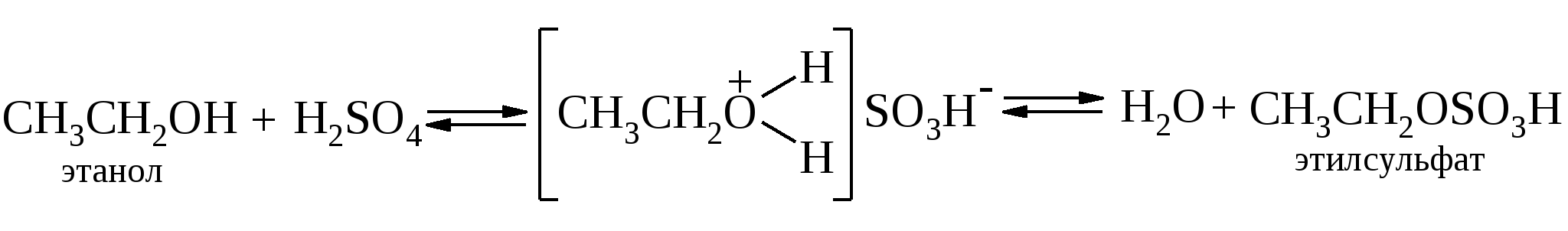
Наряду с серной и фосфорной кислотами, оксидом алюминия для дегидратации спиртов также используют щавелевую кислоту, бензолсульфокислоту, хлорид цинка и оксид тория ТhО2. Примечательно, что при нагревании вторичных спиртов с оксидом тория(IV) получаются алкены с терминальной (концевой) двойной связью. Наряду с образованием алкенов в зависимости от условий реакции (температура и концентрация кислоты) спирты могут превращаться в простые эфиры, о которых речь пойдет в соответствующей главе.
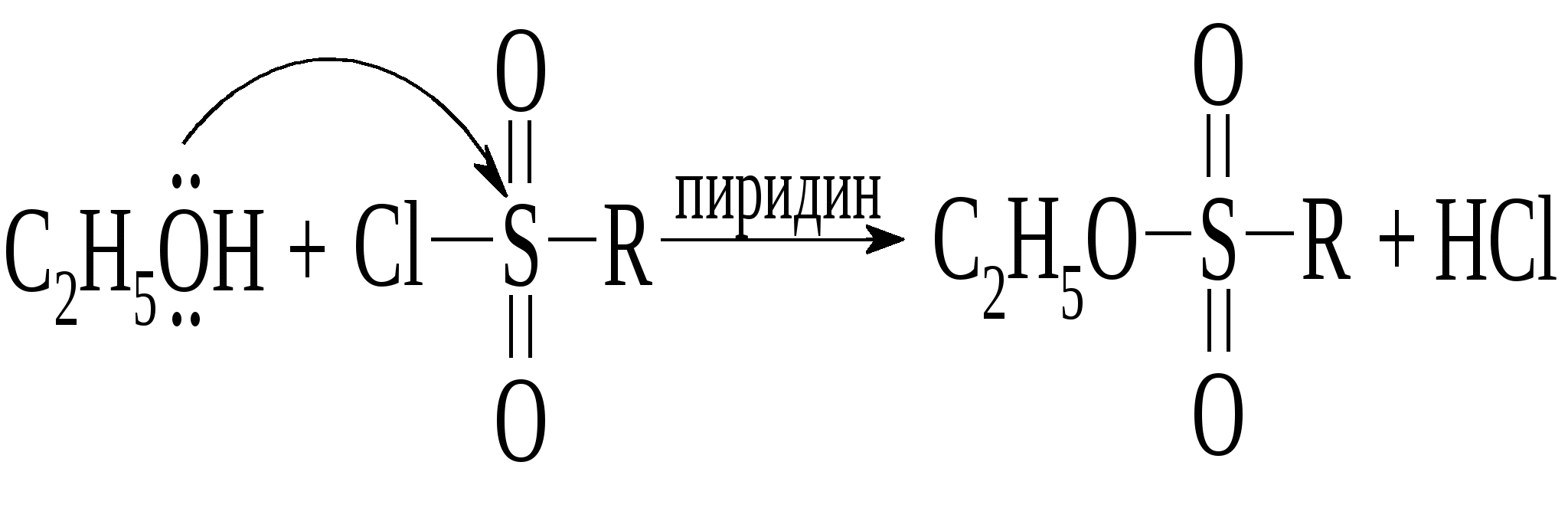 Наиболее часто применяют хлорангидриды толуолсульфокислоты, метансульфокислоты и трифторметансульфокислоты:
Эфиры сульфокислот подходящие соединения для различных нуклеофильных реакций, т.к. сульфонатная группа легко, часто при комнатной температуре, подвергается замещению, особенно это относится к «трифлатам» R-O-SO2CF3. Реакции протекают стереоспецифически с обращением конфигурации. 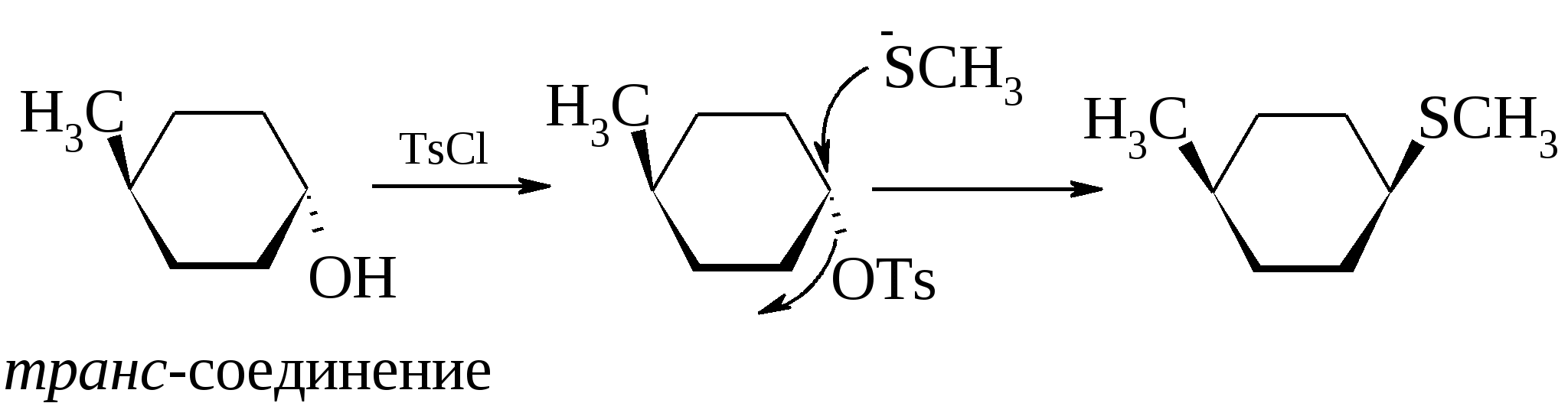
В зависимости от соотношения реагентов могут быть получены первичные, вторичные и третичные амины, а также четвертичные аммониевые соли. Использование в качестве катализатора оксида алюминия при 300 ºС приводит к таким же результатам. 13.5.2. Разрыв связи О–Н
СН3ОН > первичный > вторичный > третичный. При введении электроноакцепторных заместителей кислотность спиртов увеличивается, и, например, спирт (СF3)3СОН сравним по кислотности с карбоновыми кислотами.
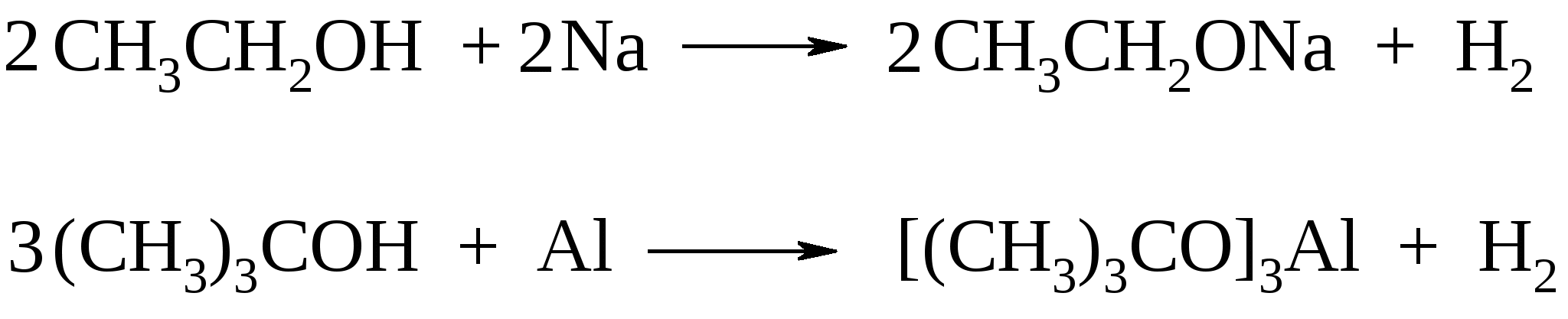
СH3CH2OH + NaNH2 → CH3CH2ONa + NH3 CH3OH + CH3MgI → CH3OMgI + CH4↑ Последняя реакция используется для количественного определения подвижных атомов водорода. Она известна под названием реакции Чугаева–Церевитинова–Терентьева. Спирты по кислотности значительно уступают воде, поэтому даже при действии концентрированных щелочей равновесие сдвинуто влево. Тем не менее, этой реакцией иногда пользуются в промышленности для получения алкоголятов простейших спиртов. С этой целью в реакционную смесь добавляют бензол, позволяющий удалять воду в виде азеотропной смеси. Среди алкоголятов спиртов следует назвать изопропилат (i-PrO)3Al и трет-бутилат (t BuO)3Al алюминия, которые служат реагентами для окисления по Оппенауэру и восстановления по Мейервейну–Понндорфу.
Окисление первичных и вторичных спиртов до альдегидов или кетонов осуществляют с помощью следующих реагентов: KMnO4, K2Cr2O7, CrO3, MnO2, Ag2O, Ag2CO3 и т.д. С бихроматом калия реакция протекает по уравнению: 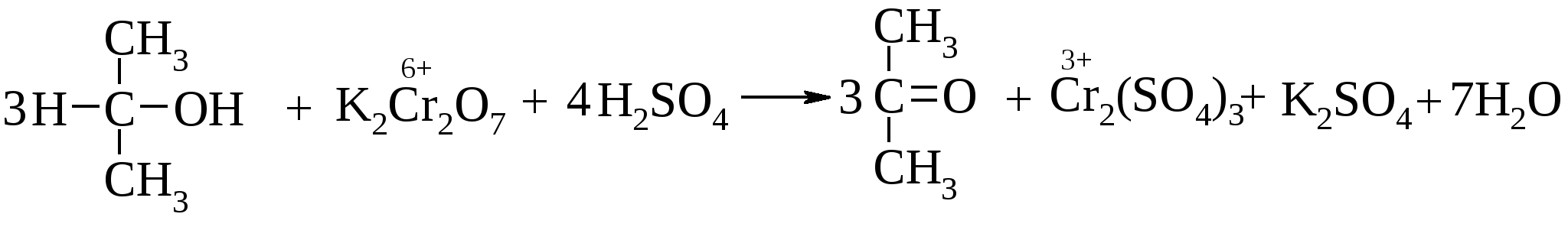 . .Установлен следующий механизм реакции: 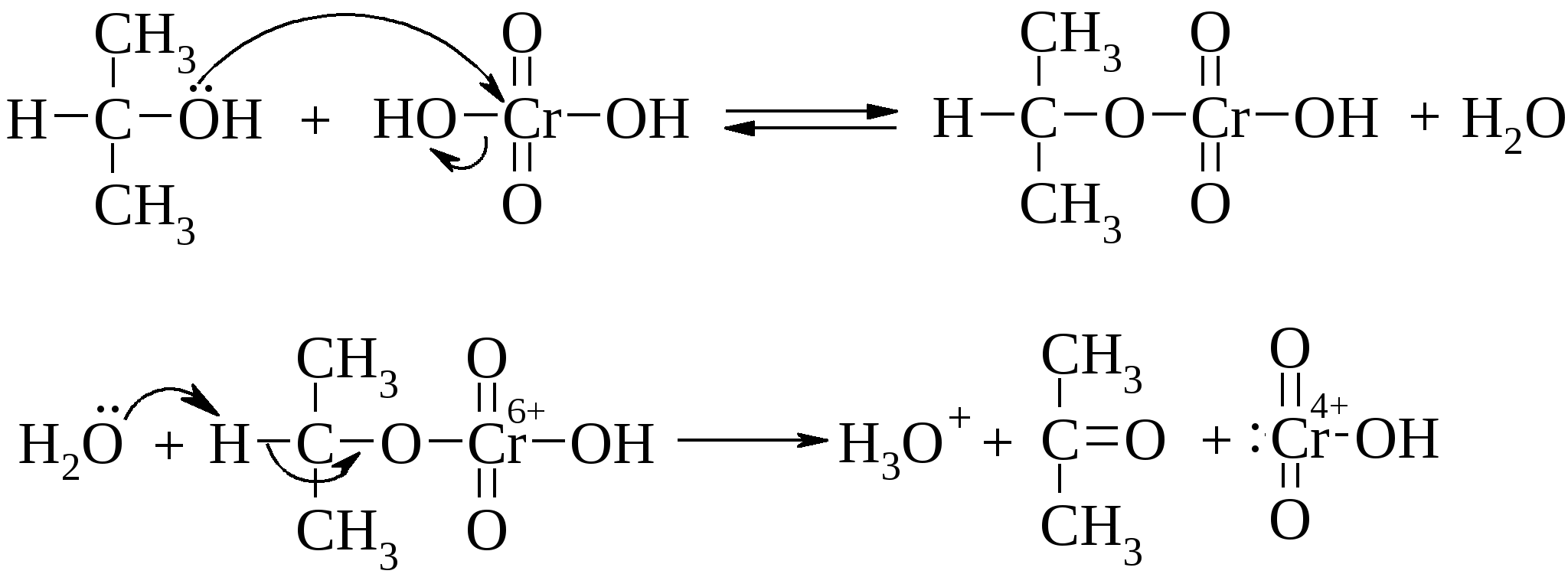 В то время как окисление вторичных спиртов останавливается на стадии получения кетонов, первичные спирты в этих условиях превращаются в альдегиды, которые через гидратную форму окисляются до карбоновых кислот: 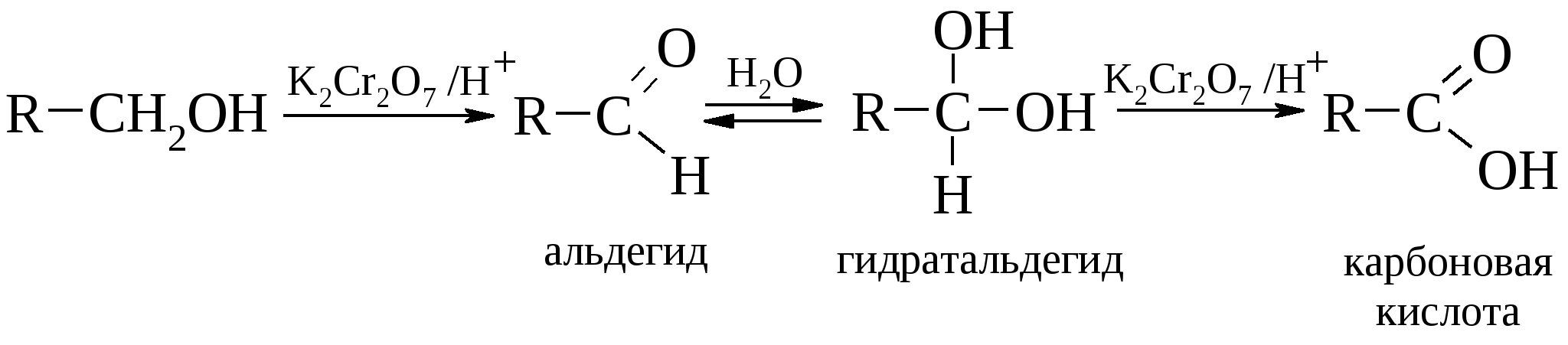 Если же есть необходимость остановить реакцию на стадии альдегида, то процесс ведут в безводном хлористом метилене. В этом случае невозможно образование гидрата альдегида, и следовательно, карбоновая кислота не синтезируется. Окисление спиртов бихроматом калия сопровождается изменением желтой окраски раствора хрома (Cr6+) на зеленую (Cr3+) и может служить в качестве контроля за протеканием реакции. Третичные спирты в обычных условиях не окисляются, однако в кислой среде они могут подвергаться дегидратации до алкенов, которые затем окисляются с деструкцией углеродной цепи. 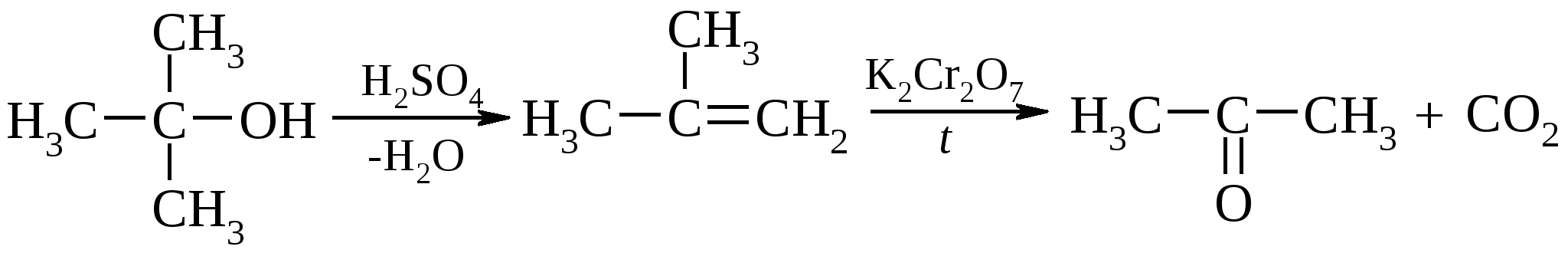
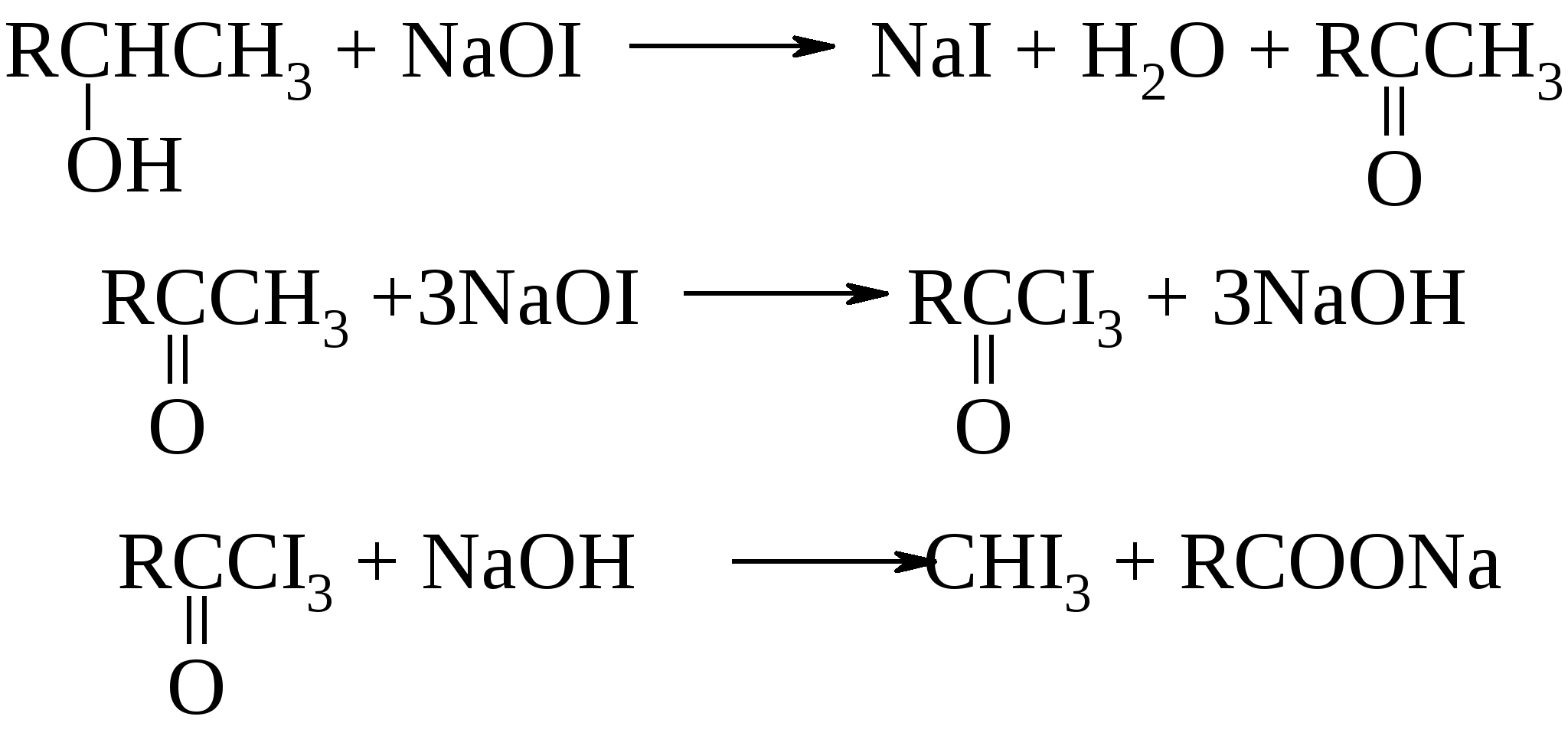 |