Зачем врачу нужна биологическая химия
 Скачать 6.47 Mb. Скачать 6.47 Mb.
|

Холестерол жизненно необходим клеткам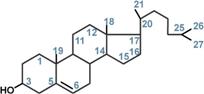 Холестерол относится к группе соединений, имеющих в своей основе циклопентан-пергидрофенантреновое кольцо, и является ненасыщенным спиртом. ИсточникиСинтезхолестерола в организме составляет примерно 0,5-0,8 г/сут, при этом половина образуется в печени, около 15% в кишечнике, оставшаяся часть в любых клетках, не утративших ядро. Таким образом, все клетки организма способны синтезировать холестерол. Из пищевых продуктов наиболее богаты холестеролом (в пересчете на 100 г продукта) сметана (0,002 г), сливочное масло (0,03 г), яйца (0,18 г), говяжья печень (0,44 г). В целом за сутки с обычным рационом поступает около 0,4 г. Выведение из организмаВыведение холестерола из организма происходит в основном через кишечник:
Функции холестерола1. Структурная– входит в состав мембран, обуславливая их вязкость и жесткость. 2. Связывание и транспортполиненасыщенных жирных кислот между органами и тканями в составе липопротеинов низкой и высокой плотности. Примерно 1/4 часть всего холестерола в организмеэтерифицирована олеиновой кислотой и полиненасыщенными жирными кислотами. В плазме крови соотношение эфиров холестерола к свободному холестеролу составляет 2:1. 3. Является предшественником желчных кислот, стероидных гормонов (кортизола, альдостерона, половых гормонов) и витамина D. Болезни накопления липидов называются липидозыГенетические заболевания, при которых происходит неполное расщепление полимерных веществ и их накопление, получили название лизосомные болезни накопления, так как они обусловлены дефектами специфических лизосомальных гидролаз. В случае накопления липидов такие болезни называютсялипидозы. При липидозах нарушается нормальный катаболизм липидов до соответствующих мономеров. ЛипидозыБолезнь Вольмана – редкое аутосомно-рецессивное заболевание из-за дефекта кислой эстеразы лизосом, что обусловливает накопление эфиров холестерола в лизосомах печени, селезенки, надпочечников, костного мозга и тонкого кишечника. Проявляется в первые недели жизни рвотой, диареей и стеатореей, гепатоспленомегалией и двусторонним кальцинозом надпочечников. Больные умирают в возрасте до 6 мес. Болезнь Шюллера-Кристиана аутосомно-рецессивное заболевание характеризуется отложением в плоских костях, твердой мозговой оболочке и коже холестерола и его эфиров. Характерными являются деструктивные изменения в костях, остеопороз, мозжечковые расстройства. Заболевание обычно начинается в возрасте до 10 лет, реже позднее. Мужчины болеют в 2 раза чаще, чем женщины. Течение заболевания прогрессирующее. Дефектный фермент неизвестен. Болезнь Гоше– отложение цереброзидовв макрофагальных клетках селезенки, печени, лимфатических узлов и костного мозга. Возникает в связи с аутосомно-рецессивным отсутствием гликоцереброзидазы (β-глюкозидазы). Основными симптомами заболевания являются спленомегалия, увеличение печени и селезенки, а также изменения в костях, проявляющиеся в виде остеопороза.  Дефектный фермент при болезни ГошеПри болезни Нимана-Пика наблюдается отложение сфингомиелинав клетках различных органов из-за дефицита сфингомиелиназы. Болезнь наследуется аутосомно-рецессивно, проявляется резким увеличением печени и селезенки, замедлением психического развития ребенка, появлением слепоты и глухоты. Чаще всего дети погибают в возрасте до 2 лет.  Дефектный фермент при болезни Нимана-ПикаБолезнь Тея-Сакса (амавротическая семейная идиотия) является результатом дефектаN-ацетилгексозаминидазы, при котором происходит отложение ганглиозидовв клетках головного мозга, что сопровождается атрофией зрительных нервов, слепотой, слабоумием и смертью в младенческом возрасте. 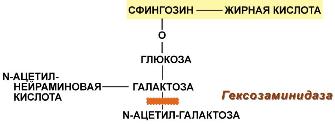 Дефектный фермент при болезни Тея-СаксаПереваривание жиров включает 5 этапов Потребность в липидах взрослого организма составляет 80-100 г в сутки, из них растительных (жидких) жиров должно быть не менее 30%. С пищей в основном поступают триацилглицеролы, фосфолипиды и эфиры ХС. Переваривание липидов осложняется тем, что их молекулы полностью или частично гидрофобны. Для преодоления этой помехи используется процесс эмульгирования, когда гидрофобные молекулы (ТАГ, эфиры ХС) или гидрофобные части молекул (ФЛ, ХС) погружаются внутрь мицеллы, а гидрофильные остаются на поверхности, обращенной к водной фазе. Условно внешний обмен липидов можно подразделить на следующие этапы: 1. Эмульгирование жиров пищи – необходимо для того, чтобы ферменты ЖКТ смогли начать работу. 2. Гидролиз триацилглицеролов, фосфолипидов и эфиров ХС под влиянием ферментов ЖКТ. 3. Образование мицелл из продуктов переваривания (жирных кислот, МАГ, холестерола). 4. Всасывание образованных мицелл в эпителий кишечника. 5. Ресинтез триацилглицеролов, фосфолипидов и эфиров ХС в энтероцитах. После ресинтеза липидов в кишечнике они собираются в транспортные формы – хиломикроны (основные) и липопротеины высокой плотности (ЛПВП) (малое количество) – и разносятся по организму. |