шпоры фх. 1. Физическая химия цель, задачи, методы исследования. Основные понятия физической химии. Физ химия
 Скачать 0.58 Mb. Скачать 0.58 Mb.
|

|
Энтропия – величина экстенсивная; она зависит от количества вещества в системе. Энтропия подчиняется закону аддитивности, т.е. энтропия равновесной системы равна сумме энтропий отдельных ее частей, а изменение энтропии всей системы равно сумме изменений энтропии ее частей. Изменение энтропии в сложном процессе равно сумме изменений энтропии в отдельных стадиях процесса. Для необратимых процессов изменение энтропии связано с теплотой процесса неравенствами. Поэтому по данным для необратимых процессов энтропию нельзя вычислить. Но изменение энтропии в обратимом и необратимом процессах одинаково, так как энтропия является функцией состояния. Следовательно, чтобы вычислить изменение энтропии в данном реальном необратимом процессе, нужно этот процесс разбить на стадии, которые проводятся обратимо, и вычислить для них изменения энтропии по уравнениям для обратимых процессов. Просуммировав изменения энтропии всех стадий, получим изменение энтропии в необратимом процессе. Вычислим изменение энтропии для разнчх процессов: 1.Для изотермического процесса (Т=const) с любым веществом. Проведем мысленно данный изотермический процесс обратимо и рассчитаем изменение энтропии по уравнению 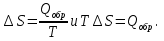 Например, для фазового перехода Например, для фазового перехода 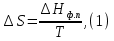 где где 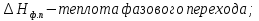 Т – абсолютная температура фазового перехода. В общем случае обратимого процесса при постоянном объеме, когда изменение внутренней энергии равно Т – абсолютная температура фазового перехода. В общем случае обратимого процесса при постоянном объеме, когда изменение внутренней энергии равно 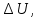 или при постоянном давлении, когда изменение энтальпии равно или при постоянном давлении, когда изменение энтальпии равно 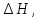 изменение энтропии равно: изменение энтропии равно: 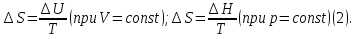 2. При нагревании любого вещества от  при постоянном объеме (V=const). В случае, как следует из математической формулироквки первого начала термодинамики, теплота процесса приобретает свойства функции состояния и не зависит от пути процесса. Подставляя значение при постоянном объеме (V=const). В случае, как следует из математической формулироквки первого начала термодинамики, теплота процесса приобретает свойства функции состояния и не зависит от пути процесса. Подставляя значение  из уравнения из уравнения 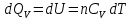 и и 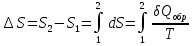 , получаем: , получаем: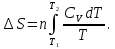 Если принять Если принять 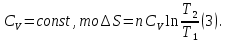 3. При нагревании любого вещества при постоянном давлении (p=const) 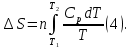 При При 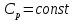 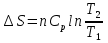 (4) (4)4.Для различных процессов с идеальным газом в соответствии с 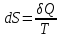 получим выражение получим выражение 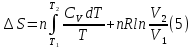 Если 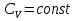 , вместо уравнения (5) можно написать , вместо уравнения (5) можно написать 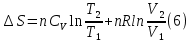 Учитывая, что для идеального газа 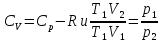 , уравнение (6) можно представить в виде: , уравнение (6) можно представить в виде: 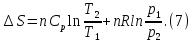 В изотермическом процессе, учитывая, что 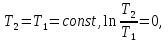 получаем из (6) и (7) получаем из (6) и (7)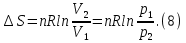 В изохорном процессе, учитывая, что 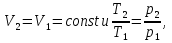 получаем из равенства (6) получаем из равенства (6)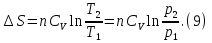 В изобарном процессе, учитывая, что 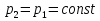 и и 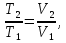 находим из (7) находим из (7)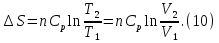 Определим изменение энтропии при взаимной диффузии двух идеальных газов. Представим себе два идеальных газа (1 и 2), находящихся в сосуде, разделенном перегородкой с отверстием. Пусть вначале в одной части сосуда объемом 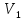 находится находится 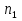 молей газа 1-го сорта, а в другой части сосуда объемом молей газа 1-го сорта, а в другой части сосуда объемом 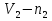 молей газа 2-го сорта, причем, давление p и температура Т обоих газов одинаковы. Газы будут взаимно диффундировать через отверстие в перегородке и через некоторое время в обеих частях сосуда образуется равномерная смесь газов; при этом молей газа 2-го сорта, причем, давление p и температура Т обоих газов одинаковы. Газы будут взаимно диффундировать через отверстие в перегородке и через некоторое время в обеих частях сосуда образуется равномерная смесь газов; при этом 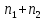 молей газов займут весь объем молей газов займут весь объем 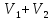 сосуда. сосуда.13. Расчет изменения энтропии реакции при стандартной и нестандартной температурах (на примере реакций с участием неорганических веществ) Термодинамическая энтропия S, часто просто именуемая энтропия, в химии и термодинамике является функцией состояния термодинамической системы. 1) теплота не может самопроизвольно переходить от менее нагретого тела к более нагретому 2) невозможен процесс, единственным результатом которого является превращение теплоты в работу Энтропия как критерий направления процессов. В изолированной системе протекают только процессы, в которых энтропия увеличивается. Энтропия есть мера упорядоченности системы. Цикл Карно: состоит из четырех участков и изображается PV-диаграммами. Процессы 1-2, 3-4 явл-ся изотерм., процессы 2-3, 4-1 явл-ся адиабатическими. 1-2: Пусть газ нах-ся в точке 1 с темп Т1, давл Р1 и объемом V1. Про изотерм. Расширении газ преходит в состояние 2 с давл Р2, V2, но Т1. Темп не меняется, значит не меняется внутр. Энергия. Чтобы газ изотерм. Расширялся, он должен получить от нагр Q1. При изотрем. Расширении Q1=W1 2-3: из сост 2 газ адиабатически расширяется до сост 3. Р3, V3, Т2 3-4: из сост 3газ изотерм сжимается до сост 4, характеризуемого параметрами V4, P4, T2. Над газом совершается работа при данном изотерм проц ΔU=0 4-1: из сост 4 газ адиабатно сжимается так, что он принимает исх парметры P1, V1, T1. Над газом совершается работа, газ нагревается до T1, т.к. теплоотдачи при адиабатическом проц нет –ΔW=ΔU Уравнения для расчета энтропии для различных систем:
Для различных процессов с идеальным газом При ΔCv=const ΔS=nCvln 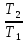 + nRln + nRln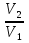 Если Cp=Cv-R ΔS=nCpln + nRln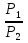 В изотермическом процессе ΔS=nCpln = nRlnВ изобарном процессе ΔS=nCpln = nRlnВ изохорном процессе ΔS=nCpln = nRlnРасчет изменения энтропии хим. Р-ции 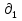 А1 + А1 + 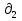 А2 = А2 = 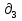 А3 + А3 + 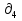 А4 А4Т=298 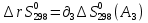 + + 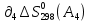 - - 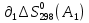 - - 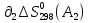 T≠298 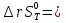 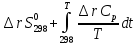
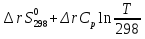
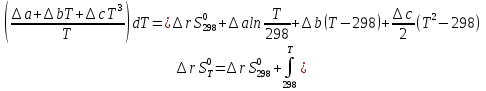 для неорг. Вв для неорг. Вв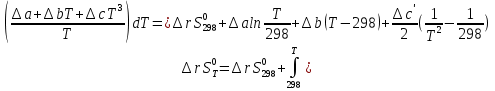 для орг. В-в для орг. В-в14.Изохорно-изотермический потенциал, его свойства, применение в качестве критерия направленности процесса. Термодинамическими потенциалами, или характеристическими функциями, называют термодинамические функции, посредством которых и их производных по соответствующим независимым переменным (естественным) могут быть выражены в явном виде все термодинамические свойства си-мы. Это означает, что характеристические функции содержат в себе всю термодинамическую информацию о си-ме. Наибольшее значение имеют четыре основных термодинамических потенциала (Пав): 1) внутренняя энергия U(S,V), 2)Энтальпия H(S,p)=U+pV, 3)энергия ГельмгольцаF(T,V)=U-TS, 4)энергия ГиббсаG(T,p)=H-TS=F+pV. В скобках указаны естественные переменные для термодинамических потенциалов. Все эти потенциалы имеют размерность энергии и все они не имеют абсолютного значения, поскольку определены с точностью до постоянной, которая равна внутренней энергии при абсолютном нуле. ϬQ=dU+ϬA, ϬA=pdV+ϬA', ϬQ=dU+pdV+δA' – первый закон термодинамики. 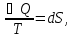 ϬQ=TdS – второй закон термодинамики. ϬQ=TdS – второй закон термодинамики.Зависимость термодинамических потенциалов от их естественных переменных описывается основным уравнением термодинамики, которое объединяет первое и второе начала: TdS=dU+ PdV=ϬA' => A'max= TdS-dU- PdV. Рассмотрим Аmax при V=const, T=const A’max= TdS – dU = -d(U - TS) U - TS=F – свободная энергия Гельмгольца A’max= -dF – изохорно-изотермический потенциал В зависимости от условия протекания процесса различают ; термодинамических потенциала  ПA,B ПA,BA'max=-ПАВ- т.е во всех случаях A'max= убыли соответствующих термодинамических потенциалов. A'max=-ПАВ- для обратимых процессов, A'max<-ПАВ- для необратимых процессов, A'max>=-ПАВ- для любых процессов. A'max=0, т.е си-ма находится под влиянием давления. ∆ПАВ<0 при любом самопроизвольном процессе термодинамический потенциал уменьшается и его можно использовать, как критерий направления процесса:
15. Изобарно-изоэнтропийный потенциал, его свойства, применение в качестве критерия направленности процесса. Термодинамическими потенциалами, или характеристическими функциями, называют термодинамические функции, посредством которых и их производных по соответствующим независимым переменным (естественным) могут быть выражены в явном виде все термодинамические свойства си-мы. Это означает, что характеристические функции содержат в себе всю термодинамическую информацию о си-ме. Наибольшее значение имеют четыре основных термодинамических потенциала (Пав): 1) внутренняя энергия U(S,V), 2)Энтальпия H(S,p)=U+pV, 3)энергия ГельмгольцаF(T,V)=U-TS, 4)энергия ГиббсаG(T,p)=H-TS=F+pV. В скобках указаны естественные переменные для термодинамических потенциалов. Все эти потенциалы имеют размерность энергии и все они не имеют абсолютного значения, поскольку определены с точностью до постоянной, которая равна внутренней энергии при абсолютном нуле. ϬQ=dU+ϬA, ϬA=pdV+ϬA', ϬQ=dU+pdV+δA' – первый закон термодинамики. ϬQ=TdS – второй закон термодинамики.Зависимость термодинамических потенциалов от их естественных переменных описывается основным уравнением термодинамики, которое объединяет первое и второе начала: TdS=dU+ PdV=ϬA' => A'max= TdS-dU- PdV. Рассмотрим Amax при P=const, S=const A’max = -dU – PdV = -d(U-PV) = -dH A’max = -dH – изобарно-изоэнтропийный потенциал 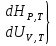 тепловой эффект тепловой эффект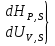 энтальпия энтальпияВ зависимости от условия протекания процесса различают ; термодинамических потенциала ПA,BA'max=-ПАВ- т.е во всех случаях A'max= убыли соответствующих термодинамических потенциалов. A'max=-ПАВ- для обратимых процессов, A'max<-ПАВ- для необратимых процессов, A'max>=-ПАВ- для любых процессов. A'max=0, т.е си-ма находится под влиянием давления. ∆ПАВ<0 при любом самопроизвольном процессе термодинамический потенциал уменьшается и его можно использовать, как критерий направления процесса:
16) Изобарно-изотермический потенциал, его свойства, применение в качестве критерия направленности процесса Термодинамическими потенциалами, или характеристическими функциями, называют термодинамические функции, посредством которых и их производных по соответствующим независимым переменным (естественным) могут быть выражены в явном виде все термодинамические свойства си-мы. Это означает, что характеристические функции содержат в себе всю термодинамическую информацию о си-ме. Наибольшее значение имеют четыре основных термодинамических потенциала (Пав): 1) внутренняя энергия U(S,V), 2)Энтальпия H(S,p)=U+pV, 3)энергия ГельмгольцаF(T,V)=U-TS, 4)энергия ГиббсаG(T,p)=H-TS=F+pV. В скобках указаны естественные переменные для термодинамических потенциалов. Все эти потенциалы имеют размерность энергии и все они не имеют абсолютного значения, поскольку определены с точностью до постоянной, которая равна внутренней энергии при абсолютном нуле. ϬQ=dU+ϬA, ϬA=pdV+ϬA', ϬQ=dU+pdV+δA' – первый закон термодинамики. ϬQ=TdS – второй закон термодинамики.Зависимость термодинамических потенциалов от их естественных переменных описывается основным уравнением термодинамики, которое объединяет первое и второе начала: TdS=dU+ PdV=ϬA' => A'max= TdS-dU- PdV. Рассмотрим Аmax при P=const, T=const A’max= TdS – dU - PdV= -d(TS – U + PV) TS –U + PV=G – свободная энергия Гиббса A’max= -dG – изобарно-изотермический потенциал В зависимости от условия протекания процесса различают ; термодинамических потенциала ПA,BA'max=-ПАВ- т.е во всех случаях A'max= убыли соответствующих термодинамических потенциалов. A'max=-ПАВ- для обратимых процессов, A'max<-ПАВ- для необратимых процессов, A'max>=-ПАВ- для любых процессов. A'max=0, т.е си-ма находится под влиянием давления. ∆ПАВ<0 при любом самопроизвольном процессе термодинамический потенциал уменьшается и его можно использовать, как критерий направления процесса:
17. Изохорно-изоэнтропийный потенциал, его свойства, применение в качестве критерия направленности процесса. Термодинамическими потенциалами, или характеристическими функциями, называют термодинамические функции, посредством которых и их производных по соответствующим независимым переменным (естественным) могут быть выражены в явном виде все термодинамические свойства си-мы. Это означает, что характеристические функции содержат в себе всю термодинамическую информацию о си-ме. Наибольшее значение имеют четыре основных термодинамических потенциала (Пав): 1) внутренняя энергия U(S,V), 2)Энтальпия H(S,p)=U+pV, 3)энергия ГельмгольцаF(T,V)=U-TS, 4)энергия ГиббсаG(T,p)=H-TS=F+pV. В скобках указаны естественные переменные для термодинамических потенциалов. Все эти потенциалы имеют размерность энергии и все они не имеют абсолютного значения, поскольку определены с точностью до постоянной, которая равна внутренней энергии при абсолютном нуле. ϬQ=dU+ϬA, ϬA=pdV+ϬA', ϬQ=dU+pdV+δA' – первый закон термодинамики. ϬQ=TdS – второй закон термодинамики.Зависимость термодинамических потенциалов от их естественных переменных описывается основным уравнением термодинамики, которое объединяет первое и второе начала: TdS=dU+ PdV=ϬA' => A'max= TdS-dU- PdV. Рассмотрим максимальную работу при V=const, S=const: A'max= - dU- это и есть изохорно изоэнтропийный потенциал. В данном случае A'max является функцией состояния, т.е не зависит от пути процесса. Величина A'max определяется уменьшением внутренней энергии си-мы. A'max=-ПАВ- т.е во всех случаях A'max= убыли соответствующих термодинамических потенциалов. A'max=-ПАВ- для обратимых процессов, A'max<-ПАВ- для необратимых процессов, A'max>=-ПАВ- для любых процессов. A'max=0, т.е си-ма находится под влиянием давления. ∆ПАВ<0 при любом самопроизвольном процессе термодинамический потенциал уменьшается и его можно использовать, как критерий направления процесса:
17. Изохорно-изоэнтропийный потенциал, его свойства, применение в качестве критерия направленности процесса. Термодинамическими потенциалами, или характеристическими функциями, называют термодинамические функции, посредством которых и их производных по соответствующим независимым переменным (естественным) могут быть выражены в явном виде все термодинамические свойства си-мы. Это означает, что характеристические функции содержат в себе всю термодинамическую информацию о си-ме. Наибольшее значение имеют четыре основных термодинамических потенциала (Пав): 1) внутренняя энергия U(S,V), 2)Энтальпия H(S,p)=U+pV, 3)энергия ГельмгольцаF(T,V)=U-TS, 4)энергия ГиббсаG(T,p)=H-TS=F+pV. В скобках указаны естественные переменные для термодинамических потенциалов. Все эти потенциалы имеют размерность энергии и все они не имеют абсолютного значения, поскольку определены с точностью до постоянной, которая равна внутренней энергии при абсолютном нуле. ϬQ=dU+ϬA, ϬA=pdV+ϬA', ϬQ=dU+pdV+δA' – первый закон термодинамики. ϬQ=TdS – второй закон термодинамики.Зависимость термодинамических потенциалов от их естественных переменных описывается основным уравнением термодинамики, которое объединяет первое и второе начала: TdS=dU+ PdV=ϬA' => A'max= TdS-dU- PdV. Рассмотрим максимальную работу при V=const, S=const: A'max= - dU- это и есть изохорно изоэнтропийный потенциал. В данном случае A'max является функцией состояния, т.е не зависит от пути процесса. Величина A'max определяется уменьшением внутренней энергии си-мы. A'max=-ПАВ- т.е во всех случаях A'max= убыли соответствующих термодинамических потенциалов. A'max=-ПАВ- для обратимых процессов, A'max<-ПАВ- для необратимых процессов, A'max>=-ПАВ- для любых процессов. A'max=0, т.е си-ма находится под влиянием давления. ∆ПАВ<0 при любом самопроизвольном процессе термодинамический потенциал уменьшается и его можно использовать, как критерий направления процесса:
18) Уравнение Гиббса – Гельмгольца. Определение изменения энергии Гиббса реакции при не стандартной температуре. Характеристической функцией называется термодинамическая функция, посредством которой или ее производных могут быть выражены в явном виде термодинамические свойства си-мы (p,V,T,S и др.). Рассмотрим энергию Гиббса как функцию температуры и давления: G=f(T,p), а энергию Гельмгольца как функцию температуры и объема: F=f(T,V). Выразим полный дифференциал функции G и F через частные производные: dG≤-SdT+Vdp (1) dF≤-SdT-pdV (2) dG= 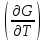 PdT+ PdT+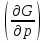 Tdp; (3) Tdp; (3)dF= 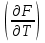 VdT+ VdT+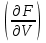 TdV. (4) TdV. (4)Сравнивая уравнения (1) с (3) и (2) с (4) для состояния равновесия си-мы, получаем P=-S; T=V; (5)V=-S; T=-p (6)Т.о, частные производные энергии Гиббса по температуре и давлению и частные производные энергии Гельмгольца по температуре и объему равны параметрам S,V или p, т.е эти функции являются характеристическими функциями. Равенства (5) и (6) позволяют вывести ряд важных уравнений химической термодинамики. Приращение энергии Гиббса или энергии Гельмгольца выражается равенствами: ∆ G=∆H-T∆S; (7) ∆F=∆U-T∆S; (8) Тогда соответственно из (5) и (6) вытекает 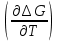 P=-∆S; (9) P=-∆S; (9)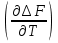 V=-∆S; (10) V=-∆S; (10)Подставляя выражения для ∆S из (9) и (10) в (7) и (8), получаем ∆ G=∆H+Т P; (11)∆F=∆U+T V; (12)Уравнения (11) и (12) называются уравнениями Гиббса-Гельмгольца. Величины ∆ G и ∆F имеют смысл максимальной работы химической реакции, когда она проводится изотермически и обратимым путем, например в электрохимическом элементе. Вторые слагаемые в правой части уравнений (11) и (12) имеют смысл теплоты обратимого процесса ,т.к -Т P=T∆S=Qобр;-Т V=T∆S=Qобр;Величины ∆H и ∆U представляют собой тепловые эффекты химических реакций или физико-химических процессов, когда они осуществляются предельно необратимо. Уравнение Гиббса-Гельмгольца используются, например, для вычисления тепловых эффектов химических реакций по опытным значениям электродвижущих сил электрохимических элементов, для вычисления ∆S и других термодинамических величин. 19) Химический потенциал, определение, условие равновесия в открытых системах. Химический потенциал идеальных и реальных систем (газы, растворы). При прохождении многих хим. процессов число молей компонентов в си-ме меняется, т.е количество компонентов в си-ме или в фазе могут быть переменными: n1n2n3…….ni ∆G=-S∆T+VdP+f(n1)+f(n2)+……f(ni) dG=f(T,P, n1n2n3…….ni) dG= P,nidT+ 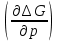 T,nidP+ T,nidP+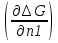 P,T,njdn1+ P,T,njdn1+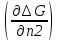 P,T,njdn2 P,T,njdn2где ni-это постоянное количество всех компонентов в си-ме, nj-это постоянное количество всех компонентов кроме одного изменение которого рассматривается. 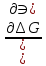 P,T,nj=ϻi P,T,nj=ϻi ϻi – химический потенциал i-го компонента. ϻi – это частная производная в данном случае энергии Гиббса по массе итого компонента при постоянных р и Т и массах остальных компонентов. ϻi – равно приращению энергии Гиббса при добавлении одного моля итого компонента к большому объему си-мы при постоянстве температуры и давления. Большой объем си-мы это когда состав си-мы почти не изменяется при добавлении 1моля компонента. ∂G= ϻi dni => G= ϻi dGT,P= ϻ1 dn1+ ϻ2 dn2+…. ϻi dni dGT,P= 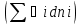 P,T , если P,T =0, то dGT,P=0 P,T , если P,T =0, то dGT,P=0это общее условие равновесие, при переменном числе компонента и постоянном р,Т. dGT,P≤0;=> P,T ≤0dU=TdS-pdV+ f(n1)+f(n2)+……f(ni) U= f(S,V, n1n2n3…….ni) dU= 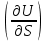 P,V,nidS+ P,V,nidS+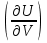 SnidV+ SnidV+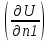 S,V,njdnj+ S,V,njdnj+ 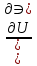 S,V,njdni S,V,njdni dH=TdS+VdP+ f(n1)+f(n2)+……f(ni) H= f(S,P, n1n2n3…….ni) dH= 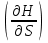 P,nidS+S,nidP+ P,nidS+S,nidP+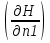 S,P,njdn1+ S,P,njdn1+ S,P,njdni. S,P,njdni.dF=-SdT-pdV+ f(n1)+f(n2)+……f(ni) F=f(T,V,n1n2….ni) dF= V,nidT+T,nidV+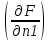 V,T,njdn1+ V,T,njdn1+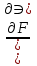 V,T,njdni. V,T,njdni.dU=TdS-PdV+ 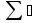 idni idnidH= TdS-VdP+ idnidF= -SdT-PdV+ idnidG= -SdT+VdP+ idniвыражает бесконечно малое изменение U,F,H,G для фазы масса которого может изменятся в результате обмена с др фазами. ϻi= S,V,njϻi= S,P,njϻi= T,V,njϻi= 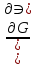 T,P,nj T,P,njХимический потенциал называется частные производные термодинамического потенциала по числу молей компонента при постоянстве естественных переменных и состава си-мы. Химический потенциал идеального и реального газа. ∂G=-SdT+VdP G=f(T,P) T=const dG=-SdT+VdP=VdP PV=nRT => V= 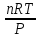 dG= dPG=nRT 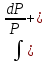 RϬ, RϬ-постоянная интегральная RϬ, RϬ-постоянная интегральнаяG=RTLnP+ RϬ (1) G0=RTLnP0+ RϬ (2) Отнимем 1ур от 2го G-G0=RT(LnP-LnP0) => G-G0=RTLn 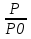 P0=1атм => G-G0=RTLnP => G=G0+RTLnP G=ϻi => ϻi = ϻi 0+RTLnP Это ур для определения химического потенциала смеси идеального газа. Льюис предложил для реальных газов использовать фугитивность( летучесть), т.к до сих пор рассматривались лишь такие си-мы газообразные части которых идеальны, т.е подчиняются уравнению PV=nRT для неидеальных систем все выше рассмотренные термодинамические соотношения не верны. Предложенное Льюисом, остается в силе и для неидеальных систем, если заменить в них фактически парциальные давления эффективными давлениями(летучесть). ϻi = ϻi 0+RTLnf, f-летучесть Летучесть совпадает с давлением если последнее настолько мало, что газ становиться идеальным. Фугитивностью(летучестью) назыв-ся величина, которую нужно подставить в выраж-е для хим-го потенциала идеального газа, чтобы получить значения реального газа. ɣ= 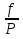 => ɣ- коэфф фугитивности , отношение фугитивности к давлению реального газа, т.е степень отклонения газов от идеального состояния. => ɣ- коэфф фугитивности , отношение фугитивности к давлению реального газа, т.е степень отклонения газов от идеального состояния.ɣmax=1, безразмерная величина. Химический потенциал для идеального и реального растворов. Для ид-го р-ра хим потенц: ϻi = ϻi 0+RTLnmi (моляльная конц) ϻi = ϻi 0+RTLnM(молярная конц) ϻi = ϻi 0+RTLnXi(мольная доля) Активность итого компонента эта величина которую нужно подставить в ур для хим потенц компонента в идеальном растворе чтобы получить действительное значение хим потенциала итого компонента в неидеальном растворе. ϻi = ϻi 0+RTLnаi ɣ= 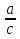 => a=ɣc => a=ɣcкоэфф. Активности- отношение активности компонента в растворе к его концентрации. ax = ɣxi Cxi(мольные доли) am= ɣmi Cmi(моляльность) aM= ɣMi CMi(молярность) 20) Химическое равновесие, вывод уравнения изотермы химической реакции. Определение стандартного значения константы равновесия реакций. Хим равновесие состояние си-мы в кот-й протекают хим реакции, но: 1) количество вещетсв не зависит от времени,2) отсутствуют потоки массы и энергии. Равновесие называют гомогенным, если реагирующие вещества наход-ся в одной фазе. И гетерогенными если они распределены по нескольким фазам. N2+3H22NH3, Ѵ1=Ѵ2( химическое равновесие) Основная задача теории хим равновесия исходя из экспериментальных (справочных) данных при произвольных температуре и давлении предсказать равновесный состав сложных смесей веществ в которых может протекать большое число хим реакций. Условия равновесия. Хим равновесие характеризуется в основном общими условиями: 1) равенство 0 всех термодинамических функций при соответствующих условиях. 2) динамическим равновесием. 3) подвижным равновесием(с изменением параметров равновесия, равновесие изменяется соответствующим образом. При прекращении действия параметров равновесия возвращается в исходное состояние. 4)Равновесие может быть достигнуто с двух сторон, со стороны увеличения или уменьшения соответствующих параметров. Основной количественный закон хим равновесия это закон действующих масс (ЗДМ) применяется для гомогенных равновесных систем. Рассмотрим в общем виде гомогенную хим реакцию. aA+bBmM+nN (1) Изменение энергии Гиббса для данной хим реакции ∆G=m(GM)+n(GN)-a(GA)-b(GB) (2) Изменение для общего i-го изменения dG(i)=VdP(i)-SidT (3) T=const, dT=0 dGi=VdPi (4) PV=RT => V=RT/P dGi=RT 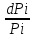 =RTlnPi (5) =RTlnPi (5)проинтегрируем ур (5)в пределах стандартного состояния газа на начала реакции (G0(i),P0i) (G(i),P0(i)) 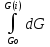 (i)= (i)=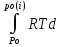 lnP(i) (6) lnP(i) (6)G(i)-G0i=RTln 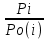 (7) (7)Давление газа в стандартном состоянии равна 1атм. G(i)=G0i+RTlnP0(i) (8) P0(i)- парциальное давление i-го участника реакции на начало реакции. С учетом ур(8), ур (2) будет выглядеть ∆G=m(G0 M)+m RTlnP0(M) +n(G0 N)+ nRTlnP0(N) -a(G0 A)-a RTlnP0(A) -b(G0 B) - bRTlnP0(B) (9) ∆G=m(G0 M)+n(G0 N)-a(G0 A)-b(G0 B) +RTln 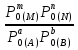 (10) (10)∆G=∆G0+RTln (11)Если в хим реакции имеет место состояния равновесия тогда ∆G будет равно 0, но парциальные давления будут иметь равновесные значения ∆G0=-RTln 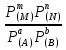 (12) (12) 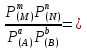 Кр (13)(константа равновесия ) Кр (13)(константа равновесия )Т.о константа равновесия любой гомогенной хим реакции протекающей в газовой фазе равна произведению равновесных парциальных давлений продуктов реакций деленному на произведение равновесных парциальных давлений исходных веществ в степенях соответствующих стехиометрическим коэф-м участников реакций. С учетом ур (13) ур (12) можно записать ∆G0=-RTlnКр (14) С учетом ур (14) ур (11) примет вид ∆G=-RTln (15)Ур (11), (15) называются ур изотермы Вант-Гоффа, еще записывается для идеального газа через парциальные давления ∆G=-RTlnКр+RTlnПр' Вывод: Величина константы равновесия зависит только от температуры и природы участников реакции. От ∆G,зависит от исходных концнтраций (активностей) или давлений( летучистей). ∆G=-RTlnКf+RTlnПf для реальных газовых систем ∆G=-RTlnКс+RTlnПс для идеальных растворов ∆G=-RTlnКm+RTlnПm ∆G=-RTlnКN+RTlnПN ∆G=-RTlnКA+RTlnA реальных растворов. Ур-е изотермы хим реакции и направление реакции. ∆G=-RTlnКр+RTlnПр'
Способы выражения константы равновесия Константа равновесия гомогенной хим реакции протекающей в газовой фазе aA+bBmM+nN (22) KP= 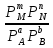 (23) (23)Поскольку ур (23) выведено из предложения , что каждый компонент хим реакции представляет собой идеальный газ. Тогда для любого участника реакции справедливо: PiV=niRT => Pi= niRT/V => ni/V=Ci => Pi=CiRT (24) Учитывая (24), ур (23) можно записать: KP= 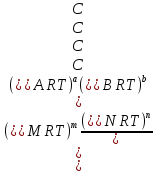 (25) (25)KP= 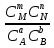 (RT)m+n-a-b== (RT)∆n (26) (RT)m+n-a-b== (RT)∆n (26)Обозначим KС= (27)Ур (27) выражает константу равновесия ур (22) выраженную через малярную концентрацию реагирующих компонентов. КР=кС(RT)∆n (28) Каждый компонент идеальной газовой смеси подчиняется закону Дальтону. Pi=XiPобщ (29) Xi- молярная доля i-го компонента газовой смеси Pобщ-общее давление газа в си-ме Подставляем ур (29) в ур (23) Кр= 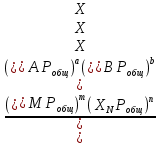 (30) (30)KP= 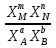 Робщ m+n-a-b= Робщ ∆n (31) Робщ m+n-a-b= Робщ ∆n (31)KX= 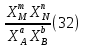 Ур (32) выражает константу равновесия через равновесные молярные доли. Сравнивая ур (32) (31) (28), дает следующие соотношения КР=кС(RT)∆n =KXPобщ∆n (33) Ур (33) выражает связь между константами равновесия для любой хим реакции которые выражены через различные равновесные концентрации, если реакция протекает без изменения числа молей газообразных участников реакции , тогда ∆n=0 КР= кС=KX (34) 21. Константа равновесия при различных способах выражения состава реакционной смеси, связь между ними. Расчет константы равновесия для реакций с изменением числа молей в системе (А+В  C и AB+C). C и AB+C). |
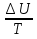 (V=const) ΔS=
(V=const) ΔS=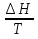 (P=const)
(P=const)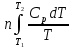 ; ΔCp=const; ΔS=nCpln
; ΔCp=const; ΔS=nCpln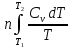 ; ΔCv=const; ΔS=nCvln
; ΔCv=const; ΔS=nCvln