шпоры фх. 1. Физическая химия цель, задачи, методы исследования. Основные понятия физической химии. Физ химия
 Скачать 0.58 Mb. Скачать 0.58 Mb.
|

|
АКТИВНОСТЬ ЭЛЕКТРОЛИТОВ В связи с электростатическим взаимодействием в растворе даже для разбавленных растворов сильных электролитов концентрации в разных уравнениях термодинамики и кинетики должны быть заменены активностями. Величина коэффициента активности определяет отклонение растворов электролитов от законов идеальных растворов, которое обусловлено в основном электростатическим взаимодействием ионов, гидратацией и другими эффектами. Для бесконечно разбавленных растворов коэффициент активности стремится к единице, уменьшаясь по мере повышения концентрации растворов. В 1927 г. Дж.Льюис и М.Рендалл установили з а к о н ион- ной силы, согласно которому "в разбавленных растворах электролитов (/ < 0,02) с одинаковой ионной силой коэффициент активности данного электролита одинаков, независимо от присутствия других электролитов". Опытное определение активности или коэффициента активности отдельно для катионов и анионов невозможно. Поэтому приходится пользоваться средними активностями или коэффициентами активности этих ионов электролита. 48. Приближения теории Дебая – Гюккеля, их концентрационные пределы применимости. где коэф. А выражается ф-лой: где В и С - эмпирич. постоянные. Ограниченность Д.-Х.т. обусловлена пренебрежением ассоциаций ионов, представлением о растворителе как о непрерывной среде, характеризуемой только значением e, т. е. неучетом мол. структуры растворителя и его взаимод. с ионами. Д.-Х. т. является основой теории электропроводности разбавл. растворов сильных электролитов, разработанной Л. Онсагером. Она позволяет объяснить увеличение электропроводности раствора при повышении напряженности постоянного электрич. поля (эффект Вина) и в высокочастотном поле (эффект Дебая-Фалькенхагена). В этих условиях ионная атмосфера, тормозящая движение ионов, не успевает образоваться. Теория создана П. Дебаем и Э. Хюккелем.Второе приближение теории Дебая-Хюккеля заключается в учете конечного размера иона a – расстояния, до которого могут сближаться центры ионов. Это приводит к изменению граничного условия и постоянной интегрирования A1: В результате основное уравнение 2-го приближения теории ДХ принимает вид: Третье приближение теории: где a, C – эмпирические константы, подбором которых можно хорошо описать эксперимент до m ≈ 1 ÷ 2. 49) Основы теории электролитической диссоциации Электролитическая диссоциация — процесс распада электролита на ионы при растворении его в полярном растворителе или при плавлении. Диссоциация на ионы в растворах происходит вследствие взаимодействия растворённого вещества с растворителем; по данным спектроскопических методов, это взаимодействие носит в значительной мере химический характер. Наряду с сольватирующей способностью молекул растворителя определённую роль в электролитической диссоциации играет также макроскопическое свойство растворителя — его диэлектрическая проницаемость. Под действием высоких температур ионы кристаллической решётки начинают совершать колебания, кинетическая энергия повышается, и наступит такой момент (при температуре плавления вещества), когда она превысит энергию взаимодействия ионов. Результатом этого является распад вещества на ионы. Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Аррениус придерживался физической теории растворов, не учитывал взаимодействие электролита с водой и считал, что в растворах находятся свободные ионы. Русские химики И. А. Каблуков и В. А. Кистяковский применили для объяснения электролитической диссоциации химическую теорию растворов Д. И. Менделеева и доказали, что при растворении электролита происходит его химическое взаимодействие с водой, в результате которого электролит диссоциирует на ионы. Для объяснения особенностей водных растворов электролитов шведским ученым С. Аррениусом в 1887 г была предложена теория электролитической диссоциации. В дальнейшем она была развита многими учеными на основе учения строения атомов и химической связи. Современное содержание этой теории можно свести к следующим трем положениям: 1) Электролиты при растворении в воде распадаются (диссоциируют) на ионы - положительные и отрицательные. Ионы находятся в более устойчивых электронных состояниях, чем атомы. Они могут состоять из одного атома - это простые ионы (Na+, Mg2+, Аl3+ и т.д.) - или из нескольких атомов - это сложные ионы (NО3-, SO2-4, РОЗ-4и т.д.). 2) Под действием электрического тока ионы приобретают направленное движение: положительно заряженные ионы движутся к катоду, отрицатель но заряженные - к аноду. Поэтому первые называются катионами, вторые - анионами. Направленное движение ионов происходит в результате притяжения их противоположно заряженными электродами. 3) Диссоциация - обратимый процесс: параллельно с распадом молекул на ионы (диссоциация) протекает процесс соединения ионов (ассоциация). Поэтому в уравнениях электролитической диссоциации вместо знака равенства ставят знак обратимости. Процесс диссоциации описывается законом действующих масс (протекает обратимо). При уменьшении концентрации диссоциация становится практически полной 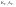 ↔ ν+ Kz+ + ν – Az- ↔ ν+ Kz+ + ν – Az-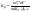 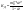 В частном случае при ν+=ν–=1 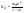 (*) (*)Здесь ν=ν++ν– - общее число ионов, образующихся при диссоциации одной молекулы, m+, m– - моляльности ионов, m(Kν+Aν–) – моляльность недиссоциированных молекул, m0 – моляльность раствора при расчете на полностью недиссоциированное вещество, α – степень диссоциации – доля диссоциированных молекул, КД – практическая константа диссоциации. 4) Коэффициент Вант-Гоффа i – изотонический коэффициент – связан со степенью электролитической диссоциации. i – среднее суммарное число частиц (ионов и молекул), образующихся при диссоциации одной молекулы i = ν + α + ν- α + (1-α) = 1 + (ν++ν–-1)α = 1 + (ν – 1)α i = 1+(ν-1)α По мере увеличения разведения коэффициент Вант-Гоффа приближается к простому целому числу (2,3,4 – в зависимости от числа ионов, образующихся при растворении молекул вещества): NaCl = Na+ + Cl- i→2 K2SO4 = 2K+ + SO  i→3 i→3AlCl3 = Al3+ + 3Cl- i→4 K4[Fe(CN)6] = 4K+ + [Fe(CN)6]2- i→5 Теория электролитической диссоциации является одной из основных теорий в неорганической химии и полностью согласуется с молекулярным учением и теорией строения атома. 50) Основные достоинства и недостатки ТЭД Аррениуса. Энергия кристаллической решетки, энергия сольватации. Электролитическая диссоциация — процесс распада электролита на ионы при растворении его в полярном растворителе или при плавлении. Диссоциация на ионы в растворах происходит вследствие взаимодействия растворённого вещества с растворителем; по данным спектроскопических методов, это взаимодействие носит в значительной мере химический характер. Наряду с сольватирующей способностью молекул растворителя определённую роль в электролитической диссоциации играет также макроскопическое свойство растворителя — его диэлектрическая проницаемость. Под действием высоких температур ионы кристаллической решётки начинают совершать колебания, кинетическая энергия повышается, и наступит такой момент (при температуре плавления вещества), когда она превысит энергию взаимодействия ионов. Результатом этого является распад вещества на ионы. Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Аррениус придерживался физической теории растворов, не учитывал взаимодействие электролита с водой и считал, что в растворах находятся свободные ионы. Русские химики И. А. Каблуков и В. А. Кистяковский применили для объяснения электролитической диссоциации химическую теорию растворов Д. И. Менделеева и доказали, что при растворении электролита происходит его химическое взаимодействие с водой, в результате которого электролит диссоциирует на ионы. Недостатки теории Аррениуса:
В теории электролитов очень важным является вопрос о распределении ионов в растворе. Распределение ионов определяется соотношением энергии электролитического взаимодействия и энергии хаотического (теплового) движения ионов. Оказывается, что эти энергии сравнимы по величине, поэтому реальное распределение ионов в электролите является промежуточным между упорядоченным и беспорядочным. Электролитические силы стремятся установить такое распределение, при котором каждый ион окружен только ионами противоположного знака, но этому противодействует хаотическое движение ионов, приводящее к беспорядочному распределению. Эти противоположные тенденции приводят к тому, что около каждого иона образуется своеобразная ионная атмосфера, в которой преобладают ионы противоположного (по сравнению с центральным ионом) знака. Аррениус предполагал, что взаимодействие ионов в растворе не влияет на их распределение и движение, которые остаются хаотическими, как и в смесях идеальных газов. Аррениус утверждал, что свойства отдельных ионов не зависят от концентрации, а некоторые свойства раствора в целом, например электропроводность, пропорциональны числу ионов. Согласно этим представлениям подвижность ионов не должна зависеть от концентрации раствора, а электропроводность сильного электролита должна возрастать с увеличением концентрации раствора. Это противоречит экспериментальным данным. По теории Аррениуса при m = 0,01 – 0,1 степень диссоциации сильных электролитов α = 0,75 – 0,95. Вычисляемые отсюда КД зависят от концентрации, то есть не являются константами. В действительности степень диссоциации сильных электролитов α→1, причем они диссоциируют необратимо. Экспериментально установлено, что недиссоциированных молекул в растворах сильных электролитов нет. Таким образом, теория Аррениуса не предусматривала деление электролитов на сильные и слабые. 51) Свойства буферных растворов, определение их рН, буферная емкость, диаграмма. Буферные растворы (англ. buffer, от buff — смягчать удар) — растворы с определённой устойчивой концентрацией водородных ионов; смесь слабой кислоты и её соли (напр., СН3СООН и CH3COONa) или слабого основания и его соли (напр., NH3 и NH4CI). Величина рН буферного раствора мало изменяется при добавлении небольших количеств свободной сильной кислоты или щёлочи, при разбавлении или концентрировании. Буферные растворы широко используют в различных химических исследованиях. Буферная ёмкость раствора — определяет способность раствора сохранять постоянной концентрацию определённых ионов (обычно применяется к ионам H+) при условии, что в растворе протекают химические реакции, или при добавлении к раствору электролитов. Равновесие в растворах электролитов для процессов диссоциации, гидролиза. Произведение растворимости труднорастворимого электролита. Буферные растворы, их свойства, буферная емкость. Произведение растворимости труднорастворимого соединения. Рассчитывается на основе закона действия масс. Можно экспериментально определить потенциометрическим и кондуктометрическим методами. При определенных условиях величина постоянная. Буферными растворами называются растворы с устойчивой концентрацией водородных ионов, т.е. с определенным значением рН, которое практически не зависит от разбавления раствора и слабо меняется при прибавлении к раствору некоторого количества сильной кислоты или сильного основания. Важной характеристикой буферной системы является зона буферного действия – интервал рН, в котором проявляется буферное действие. По составу буферные системы бывают трех типов: 1. Система из слабой кислоты и ее соли, образованной сильным основанием, например, ацетатная CH3COOH + CH3COONa, карбонатная H2CO3 + NaHCO3, белковая Pt-COOH + Pt-COONa, где Pt – протеин (белок), и др. 2. Система из слабого основания и его соли, образованной сильной кислотой, например, аммиачная NH4OH+NH4Cl. 3. Система из двух солей многоосновной кислоты с разной степенью замещенности в ее молекуле ионов водорода, например, фосфатная NaH2PO4+Na2HPO4. Рассмотрим, ацетатный буфер: CH3COOH + CH3COONa. В этом буфере [H+] будет зависеть только от степени диссоциации молекул кислоты: CH3COOH ↔ CH3COO- + H+, в общем виде НА ↔ Н+ + А- CH3COONa ↔ CH3COO- +Na+ Согласно закону действующих масс Кдис кислоты: K=[H ][CH COO ]/ [CH COOH ] → [ H ]= K[CH COOH ]/ [CH COO ] Исходя из этого выражения, уравнение можно записать в общем виде для кислотного буфера: [ H+ ]= K[ кислота ] / [ соль ] pH= - lg[H+ ]= - lg K - lg [ кислота ] / [ соль ] Тогда pH = pK + lg [ соль ] / [ кислота ] А для щелочного буфера [OH- ]= K[ основание ] / [ соль ] pOH= - lg[OH- ]= - lg K - lg [ основание ] / [ соль ] pOH= pK + lg [ соль ] / [ основание ] → pH= 14 - pOH =14 - lg K + lg [основание ] / [ соль ] Буферная емкость ß – это количество сильной кислоты или основания (в г-экв), при добавлении которого к буферному раствору рН меняется на единицу: ß= dв/ dpH = - da/ dpH, (знак (-) указывает на то, что добавление кислоты уменьшает рН. На практике буферную емкость раствора можно определить из анализа кривых титрования зависимости рН раствора от количества прибавленной к нему щелочи или кислоты. 52) Определение рН гидратообразования и произведения растворимости гирооксидов металлов. рН гидратообразования - минимальное значение рН, при котором этот осадок начинает образовываться. Раствор любой соли обладает определенной величиной рН, зависящей от равновесия гидролиза. Уменьшение кислотности, например, за счет добавления щелочи вызывает увеличение рН до определенной величины, при которой начинается выпадение осадка малорастворимой гидроокиси металла или его основной соли. В процессах водоподготовки и водоочистки очень часто важно знать рН, при котором начинается образование осадков. Зная ПР веществ, которые могут выпасть в осадок, можно подсчитать величину рН начала гидратообразования (табл.). Таблица. Определение рН растворов при гидролизе солей
Концентрация водородных ионов, соответствующая началу выпадения гидроксида, в зависимости от концентрации металла Смет и произведения растворимости гидроксида ПРмет(ОН)z выражается уравнением рН = 14 +1/zПРмет(ОН)z - 1/zlgCмет. Из выражения видно, что увеличение концентрации соли вызывает повышение концентрации ионов металла и сдвигает рН гидратообразования в сторону меньших значений, т.е. в область более кислых растворов. Рассмотрим процесс гидролиза, протекающий около поверхности растворяющегося стального анода в растворах различных солей, образованных сильным основанием и сильной кислотой (реакция среды водного раствора - нейтральная). Выделяющиеся ионы Fe2+ частично окисляются кислородом воздуха, растворенного в растворе, до Fe3+. Используя константу гидролиза ионов Fе2+и Fe3+ (10-9,5 и 5.103 соответственно), можно рассчитать рН среды в прианодном пространстве. При гидролизе Fe2+ Fe2+ + Н2О ←→ FeOH+ + Н+ получим
При [Fe2+] = 1 моль/л значение рН не более 4,75. Увеличение концентрации ионов Fe2+ до рН гидратообразования, сопровождающегося выпадением осадка Fe(OH)2, стабилизирует рН среды около значения 10, так как ПР(Fе(ОН2)) = [Fe2+] · [ОН-]2 = 4,8·10-16; [ОН-]= 3√4,8·10-16 =3,5 ; рОН = 3,5; рН = 14 - 3,5 = 10,5. Однако даже незначительное количество ионов Fe3+ в растворе приводит к сильному подкислению среды за счет гидролиза этих ионов Fe3++H2O ←→ FeOH2++H+,
При [Fe3+] =1 моль/л рН составляет примерно 2,0 53. удельная электропроводность растворов электролитов, зависимость от температуры и концентрации. Электрическая проводимость растворов электролитов, т.е. способность их проводить электрический ток, зависит от природы электролита и растворителя, концентрации, температуры и некоторых других факторов. Различают удельную и молярную электрическую проводимости. Удельная электрическая проводимость раствора электролита 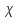 - это электрическая проводимость объёма раствора, заключённого между двумя параллельными электродами, имеющими площадь по одному квадратному метру и расположенными на расстоянии одного метра друг от друга. - это электрическая проводимость объёма раствора, заключённого между двумя параллельными электродами, имеющими площадь по одному квадратному метру и расположенными на расстоянии одного метра друг от друга.Удельная электропроводность является величиной, обратно удельному сопротивлению 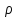 : : 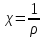 . .Удельное сопротивление определяется по уравнению 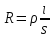 . Единица удельного сопротивления выражается величиной [ . Единица удельного сопротивления выражается величиной [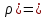 Ом*м. единица удельной электрической проводимости, выражается обратной величиной =1/(Ом*м)=Ом-1*м-1=См*м-1. Удельная электропроводность зависит от: природы электролита, температуры, давления, разведения. Ом*м. единица удельной электрической проводимости, выражается обратной величиной =1/(Ом*м)=Ом-1*м-1=См*м-1. Удельная электропроводность зависит от: природы электролита, температуры, давления, разведения. 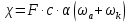 – для слабых электролитов – для слабых электролитов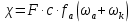 – для сильных электролитов – для сильных электролитовПовышение температуры на 1К увеличивает удельную электропроводность на 2-2,5%. Это объясняется понижением вязкости раствора и уменьшением гидратации ионов, а для растворов слабых электролитов увеличением их степени диссоциации. Зависимость удельной электропроводности разбавленных растворов от температуры описывается эмпирическим уравнением T=298[1+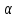 (T-298)+ (T-298)+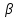 (T-298)2]. =0.0163(-0,0174), где 298 – удельная электропроводность при 298К, и - температурные коэффициенты электропроводности. Коэффициенты и зависят от природы электролита. (T-298)2]. =0.0163(-0,0174), где 298 – удельная электропроводность при 298К, и - температурные коэффициенты электропроводности. Коэффициенты и зависят от природы электролита. В растворах слабых электролитов диссоциация молекул электролита на ионы увеличивает объем раствора. Поэтому повышение давления в соответствии принципом смещения подвижного равновесия Ле-Шателье-Брауна уменьшает степень диссоциации электролита и, следовательно, электрическую проводимость. Заметное влияние на электропроводность раствора слабого электролита оказывает только давление около сотен и тысяч атмосфер. Зависимость удельной электропроводности от концентрации выражается уравнениями: 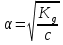 - для слабых электролитов - для слабых электролитов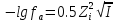 - для сильных электролитов - для сильных электролитов54. Молярная электропроводность. Закон Кольрауша. Определение молярной электропроводности при бесконечном разбавлении растворов сильных и электролитов. Электрическая проводимость растворов электролитов, т.е. способность их проводить электрический ток, зависит от природы электролита и растворителя, концентрации, температуры и некоторых других факторов. Различают удельную и молярную электрическую проводимости. Молярная электропроводность 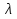 - электропроводность объема раствора электролита, содержащего 1моль-экв растворенного вещества и находящегося между двумя параллельными электродами, расположенными на расстоянии 1 метра друг от друга. Удельная и молярная электропроводности связаны между собой соотношением: =/Cm, где Cm – концентрация электролита, выраженная в моль/м3. Единица в молярной электропроводности []=См*см2*моль-1. Молярная электропроводность с уменьшением концентрации раствора увеличивается и при С - электропроводность объема раствора электролита, содержащего 1моль-экв растворенного вещества и находящегося между двумя параллельными электродами, расположенными на расстоянии 1 метра друг от друга. Удельная и молярная электропроводности связаны между собой соотношением: =/Cm, где Cm – концентрация электролита, выраженная в моль/м3. Единица в молярной электропроводности []=См*см2*моль-1. Молярная электропроводность с уменьшением концентрации раствора увеличивается и при С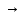 0 стремится к некоторому предельному максимальному значению 0 стремится к некоторому предельному максимальному значению 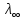 , которая называется молярной электропроводностью при предельном(бесконечном) разбавлении. Кольрауш установил, что при бесконечном разведении катионы и анионы проводят электрический ток независимо друг от друга, так как в этих условиях взаимодействие между ионами почти полностью отсутствует, в этом случае молярная электропроводность раствора будет равна сумме электропроводности катионов и анионов. Для предельно разбавленного раствора =1, поэтому = , которая называется молярной электропроводностью при предельном(бесконечном) разбавлении. Кольрауш установил, что при бесконечном разведении катионы и анионы проводят электрический ток независимо друг от друга, так как в этих условиях взаимодействие между ионами почти полностью отсутствует, в этом случае молярная электропроводность раствора будет равна сумме электропроводности катионов и анионов. Для предельно разбавленного раствора =1, поэтому =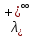 + +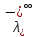 , где и - подвижности ионов при предельном разбавлении. По измеренным сопротивлениям для каждой из концентрации вычисляют удельную электропроводность, молярную электропроводность, а затем вычисляют степень диссоциации по уравнению: , где и - подвижности ионов при предельном разбавлении. По измеренным сопротивлениям для каждой из концентрации вычисляют удельную электропроводность, молярную электропроводность, а затем вычисляют степень диссоциации по уравнению: 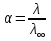 . При разбавлении слабого электролита, степень их диссоциации увеличивается, число ионов увеличивается. Для сильных электролитов зависимость молярной электропроводности от концентрации выражается эмпирическим уравнением Кольрауша: . При разбавлении слабого электролита, степень их диссоциации увеличивается, число ионов увеличивается. Для сильных электролитов зависимость молярной электропроводности от концентрации выражается эмпирическим уравнением Кольрауша: 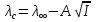 . .55. Молярная электропроводность. Влияние температуры и концентрации на молярную электропроводность растворов сильных и слабых электролитов. Молярная электропроводность - электропроводность объема раствора электролита, содержащего 1моль-экв растворенного вещества и находящегося между двумя параллельными электродами, расположенными на расстоянии 1 метра друг от друга. Удельная и молярная электропроводности связаны между собой соотношением: =/Cm, где Cm – концентрация электролита, выраженная в моль/м3. Единица в молярной электропроводности []=См*см2*моль-1. Для слабых электролитов: , - полная ионизация(=1) , - полная ионизация(=1)Для сильных электролитов:  - полное отсутствия межионного взаимодействия( - полное отсутствия межионного взаимодействия(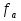 =1) =1)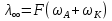 - молекулярная электропроводность при таком сильном разведении, когда слабые электролиты полностью диссоциируют на ионы, а у сильных – отсутствует межионное взаимодействие. - молекулярная электропроводность при таком сильном разведении, когда слабые электролиты полностью диссоциируют на ионы, а у сильных – отсутствует межионное взаимодействие. Зависимость молярной электропроводности от температуры можно представить уравнением: T=298[1+(T-298)], где T и298 – молярные электропроводности при температуре Т и 298К, – температурный коэффициент электропроводности. Для слабых электролитов изменение молярной электропроводности от концентрации раствора связана в основном со степенью диссоциации и для сильных электролитов – с межионным взаимодействием. Зависимость молярной электропроводности от концентрации: 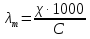 . Для сильных электролитов . Для сильных электролитов 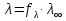 . Таким образом, в растворах сильных электролитов изменение молярной электропроводности с концентрацией обусловленной коэффициентом электропроводности, то есть влиянием электростатического взаимодействия ионов на скорость их движения. . Таким образом, в растворах сильных электролитов изменение молярной электропроводности с концентрацией обусловленной коэффициентом электропроводности, то есть влиянием электростатического взаимодействия ионов на скорость их движения. 56. Электролиз, законы электролиза. Электролиз водных растворов солей с инертным анодом (привести пример). Электролиз - окислительно-восстановительный процесс, протекающий под действием электрического тока. Законы электролиза: 1 закон: масса вещества, выделившегося на электроде, прямо пропорциональна количеству электричества, пропущенного через раствор или расплав электролита. 2 закон: если через растворы нескольких электролитов пропустить одинаковое количество электричества, то массы выделившихся веществ будут прямо пропорциональны их электрохимическим эквивалентам. 57. Определение стандартного значения электродных потенциалов. Уравнение Нернста для определения ЭДС цепей. Скачок электрического потенциала между электродом, на котором происходит окислительно-восстановительная реакция, и раствором, устанавливающийся при равенстве скоростей прямой и обратной реакций, называется равновесным потенциалом электрода в данном растворе. Причина возникновения заряда на границе металл-раствор является возникновение двойного электрического слоя.В Общем случае зависимость потенциала какого-либо электрода от состава раствора и температуры дается уравнением Нернста: 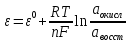 . .ЭДС элемента называется разность потенциалов на полюсах обратимого электрохимического элемента. ЭДС элемента измеряют при помощи компенсационного метода, которое заключается в том, что ЭДС вспомогательного нормального элемента сравнивается с неизвестной ЭДС. В качестве вспомогательного нормального элемента обычно применятся так называемый нормальный элемент Вестона. Этот элемент сохраняет длительное время постоянное и устойчивое значение ЭДС. ЭДС электрохимического элемента считается положительным, если электрохимическая цепь записана так, что катионы при работе элемента проходят в растворе от левого электрода к правому и в том же направлении движутся электроны по внешней цепи. При этом левый электрод является отрицательным, а правый – положительным. 58. Классификация электродов, правила записи электродов и цепей. В зависимости от природы электродной реакции различают несколько типов электродов. 1)Электроды 1го рода. Электродом первого рода называют металл или неметалл, погруженный в раствор, содержащий его ионы. Электрод первого рода можно представить в виде схемы: Мz+ |М. Ему отвечает электродная реакция: Мz+ + zе = М. Потенциал электрода первого рода можно записать: 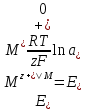 , , Где 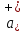 - активность ионов металла в растворе. Активность атомов в электроде из чистого металла принимается равной единице. Примером электрода 1 рода может служить медный электрод в растворе соли меди. К неметаллическим электродам первого рода относят селеновый электрод. - активность ионов металла в растворе. Активность атомов в электроде из чистого металла принимается равной единице. Примером электрода 1 рода может служить медный электрод в растворе соли меди. К неметаллическим электродам первого рода относят селеновый электрод. 2)Электроды 2 рода. Электрод второго рода состоит из металла, покрытого слоем его малорастворимого соединения и погруженного в раствор растворимой соли, содержащий тот же анион, что и малорастворимое соединение. Электрод второго рода и протекающая на нем электродная реакция записывается в виде схемы: Аz-|MA, M. Реакцию можно записать: MA + ze  M + Az-. M + Az-.Потенциал электрода второго рода можно представить выражением: 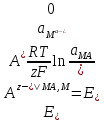 , , Электроды второго рода широко применяются в электрохимических измерениях в качестве электродов сравнения, так как их потенциал устойчив во времени и хорошо воспроизводится. Примерами электрода второго рода могут служить каломельный и хлорсеребряный электроды. 3)Газовые электроды. Газовый электрод состоит из инертного металла(обычно платины), контактирующего одновременно с газом и раствором, содержащим ионы этого газообразного вещества. Примерами газовых электродов могут служить водородный, кислородный и хлорный электроды. Газовые электроды иногда относят к электродам первого рода. Водородный электрод записывается в виде схемы: H+|H2,Pt и протекающая на нем реакция в виде H+ + e = 1/2H2. Содержание газообразного вещества, участвующего в электродной реакции, принято выражать в единицах давления чистого газа или его парциального давления в газовой смеси. Потенциал водородного электрода описывается уравнением: 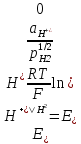 . .4)Амальгамные электроды. Амальгамный электрод состоит из амальгамы данного металла в контакте с раствором, содержащим ионы этого металла. Амальгамный электрод можно представить схемой: Mz+|M,Hg. Ему отвечает электродная реакция Mz+ + ze = M(Hg). Потенциал амальгамного электрода: 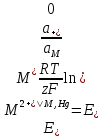 , где и , где и 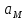 - активности ионов металла соответственно в водном растворе и амальгаме. - активности ионов металла соответственно в водном растворе и амальгаме.5)Окислительно-восстановительные электроды. Этот электрод состоит из инертного металла, погруженного в раствор, содержащий окисленную и восстановленную формы вещества. Различают простые и сложные окислительно-восстановительные системы. В простой окислительно-восстановительной системе электродная реакция состоит в изменении заряда ионов: O,R|Pt O + ze = R. Примером сложный окислительно-восстановительной системы может служить система из ионов Mn 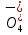 и Mn2+. и Mn2+. 59.Химические цепи(гальванический элемент), их классификация. Гальванические элементы подразделяются на три большие группы: физические, химические и концентрационные. Физические цепи – в них электроды отличаются друг от друга только физическими свойствами. Это могут быть различные модификации одного и того металла, но в разных формах устойчивости. Источником электрической энергии физических цепей служит свободная энергия перехода электрона из менее устойчивого в более устойчивое состояния. В химических цепях электроды отличаются друг от друга химическими свойствами. В этих цепях источником электрической энергии служит химическая реакция. Различают химические цепи с двумя и одним электролитом. К электрохимическим элементам с двумя электролитами относятся элемент Даниэля-Якоби. Химические цепи с одним электролитом могут быть двух видов. В цепях первого вида на одном электроде протекает реакция с участием катиона электролита, а на другом электроде с участием аниона электролита. В элементах второго вида с одним электролитом на обоих электродах протекают реакции с участием аниона электролита; при этом один электрод – газовый, а в другой – второго рода. Химические цепи с одним электролитом широко применяются в физико-химических исследованиях. Концентрационные цепи. Концентрационными цепями называют те цепи, в которых оба электрода одинаковы по своей природе, но различаются активностью одного или нескольких участников электродной реакции. При этом электрическая энергия получается за счет выравнивания концентрации веществ в элементе. Концентрационные цепи могут быть без переноса и с переносом. Концентрационными цепями без переноса называются элементы: а)с одинаковыми электродами и двумя одинаковыми по природе, но разными по концентрации растворами электролитов. Б) с электродами из двух сплавов (амальгам), одинаковых по природе, но разных по концентрации; в)с газовыми электродами, одинаковыми по природе, но с разным давлением газа на электродах. Концентрационными цепями с переносом называются элементы с одинаковыми электродами и двумя одинаковыми по природе, но разными по концентрации растворами электролитов. 60.Гальванический элемент. Термодинамика гальванического элемента. Превращение химической энергии в электрическую возможно при помощи электрохимического(гальванического) элемента, примером которого может служить элемент Даниэля-Якоби, состоящий из цинкового и медного электродов, опущенных соответственно в растворы сульфатов цинка и меди, разделенные пористой диафрагмой во избежание их перемешивании. Схема электрохимической цепи элемента Даниэля-Якоби записывается след. образом: Zn|ZnSO4||CuSO4|Cu. Вертикальной сплошной чертой обозначается граница между металлом и раствором, если на границе между двумя электролитами устранен так называемый диффузионный потенциал, то границу между двумя электролитами обозначают двумя вертикальной черточками, если не устранен, то пунктирной вертикальной чертой. Зависимость максимальной полезной работы химической реакции в гальваническом элементе от температуры можно связать с уравнениями Гиббса-Гельмгольца: G T(G/ T )р ; F U T (F / T)v. Максимальная полезная работа электрохимической реакции равна: Аmax=-G. Энтропию электрохимической реакции протекающей в гальваническом элементе определяют по формуле:  , а энтальпию , а энтальпию  или или  . .Связь константы равновесия химической реакции и стандартной E0 выражается соотношением: 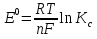 |