|
|
ШПОРЫ БХ 2 ИТОГОВАЯ. Эндергонические процессы, протекающие с увеличением свободной энергии. Катаболические превращения
Регуляция скорости β-окисления. Β-окисление-метаболический путь , прочно связанный с работой ЦПЭ и общего пути катаболизма. Поэтому его скорость регулируется потребностью клетки в энергии, т.е. соотношениями АТФ/АДФ и NADH/NAD+ , так же, как и скорость реакции ЦПЭ и общего пути катаболизма. Скорость β-окисления в тканях зависит от доступности субстрата, т.е. от количества жирных кислот, поступающих в митохондрии. Концентрация свободных жирных кислот в крови повышается при активации липолиза в жировой ткани при голодании под действием глюкагона и при физической работе под действием адреналина. В этих условиях жирные кислоты становятся преимущественным источником энергии для мышц и печени, так как в результате β-окисления образуются NADH и ацетил-КоА, ингибирующие пируватдегидрогеназный комплекс. Превращение пирувата, образующегося из глюкозы, в ацетил-КоА замедляется. Накапливаются промежуточные метаболиты гликолиза и, в частности, глюкозо-6-фосфат. Глюкозо-6-фосфат ингибирует гексокиназу и, следовательно, препятствует использованию глюкозы в процессе гликолиза. Таким образом, преимущественное использование жк как основного источника энергии в мышечной ткани и печени сберегает глюкозу для нервной ткани и эритроцитов. Скорость β-окисления также зависит от активности фермента карнитинацилтрансферазы I. В печени этот фермент ингибируется малонил КоА, веществом , образующимся при биосинтезе жк. В абсорбтивный период в печени активируется гликолиз и увеличивается образование ацетил-КоА из пирувата. Первая реакция синтеза жк-превращение ацетил-КоА в малонил-КоА. Малонил-КоА ингибирует β-окисление жк, котрые могут использоваться для синтеза жира.
Образование малонил-КоА из ацетил-КоА-регуляторная реакция в биосинтезе жк. Первая реакция синтеза жк-превращение ацетил-КоА в малонил-КоА. Фермент, катализирующий эту реакцию(ацетил Коа-карбоксилаза), относят к классу лигаз. Он содеожит ковалентно связанный биотин . В первой стадии реакции СО2 ковалентно связывается с биотином за счет энергии АТФ, во 2 стадии СОО- переносятся на ацетил-КоА с образованием малонил-КоА. Активность фермента ацетил-КоА-карбоксилазы определяет скорость всех последующих реакций синтеза жк. Поступивший в цитозоль цитрат активирует фермент ацетил-КоА-карбоксилазу. Малонил-КоА в свою очередь угнетает перенос высших жирных кислот из цитозоля в матрикс митохондрий, ингибируя активность внешней ацетил-КоА:карнитин-ацилтрансферазы, выключая таким образом окисление высших жирных кислот.
Транспорт ацил Ко-А через внутреннюю мембрану митохондрий. Так как синтез жк происходит происходит в цитозоле клеток, то ацетил-КоА должен быть транспортирован через внутреннюю мембрану митохондрий в цитозоль. Однако внутренняя мембрана митохондрий непроницаема для ацетил-КоА, поэтому в матриксе митохондрий ацетил-КоА конденсируется с оксалоацетатом с образованием цитрата при участии цитратсинтазы :
Ацетил-КоА+Оксалоацетат→ Цитрат+HS-КоА
Затем транслоказа переносит цитрат в цитоплазму. Перенос цитрата в цитоплазму происходит только при увеличении количества цитрата в митохондриях, когда изоцитратдегидрогеназа и α-кетоглутаратдегидрогеназа ингибированы высокими концентрациями NADH и АТФ. Эта ситуация создается а абсорбтивном периоде, когда клетка печени получает достаточное количество источников энергии. В цитоплазме цитрат расщепляется под действием фермента цитратлиазы :
Цитрат+HSКоА+АТФ→Ацетил-КоА+АДФ+Pi+Оксалоацетат
Ацетил-КоА в цитоплазме служит исходным субстратом в для синтеза жк, а оксалоацетат в цитозоле подвергается превращениям, в результате которых образуется пируват.
Вопрос 49.Биосинтез ТАГ (липогенез). Особенности биосинтеза ТАГ в печени и жировой ткани. Гормональная регуляция. Образование ЛПОНП в печени.
Печень - основной орган, где идет синтез жирных кислот из продуктов гликолиза. В гладком Эр гепатоцитов жирные кислоты активируются и сразу же используются для синтеза жиров, взаимодействуя с глицерол-3-фосфатом. Как и в жировой ткани, синтез жиров идет через образование фосфатидной кислоты. Синтезированные в печени жиры упаковываются в ЛПОНП и секретируются в кровь.В состав ЛПОНП, кроме жиров, входят холестерол, фосфолипиды и белок апоВ-100. Это очень «длинный» белок , содержащий 11536 аминокислот. Одна молекула апоВ-100 покрывает поверхность всего липопротеина. ЛПОНП из печени секретируется в кровь, где на них, как и на ХМ, действует ЛП-липаза. Жирные кислоты поступают в ткани, в частности в адипоциты, и используются для синтеза жиров. В процессе удаления жиров из ЛПОНП под действием ЛП-липазы ЛПОНП сначала превращаются в ЛППП , а затем в ЛПНП. В ЛПНП основными липидными компонентами служат холестерол и его эфиры, поэтому ЛПНП являются липопротеинами, доставляющими холестерол в периферические ткани. Глицерол, освободившийся из липопротеинов, кровью транспортируется в печень, где опять может использоваться для синтеза жиров. Скорость синтеза ЖК и жиров в печени существенно зависит от состава пищи. Если в пище содержится белее 10% жиров, то скорость синтеза жиров в печени резко снижается.
Гормональная регуляция синтеза жиров. Какой процесс будет преобладать в организме – синтез жиров или их распад зависит от поступления пищи и физической активности В абсорбтивном состоянии под действием инсулина происходит липогенез, в постабсорбтивном состоянии – липолиз, активируемый глюкагоном. Адреналин, секреция которого увеличивается при физической активности, также стимулирует липолиз. В абсорбтивный период при увеличении соотношения инсулин/глюкагон в печени активируется синтез жиров. В жировой ткани индуцируется синтез ЛП-липазы в адипоцитах и осуществляется ее экспонирование на поверхность эндотелия , следовательно в этот период увеличивается поступление жирных кислот в адипоциты. Одновременно инсулин активирует белки-переносчики глюкозы – ГЛЮТ-4. Поступление глюкозы в адипоциты и гликолиз также активируется. В результате образуются все необходимые компоненты для синтеза жиров: глицерол-3-фосфат и активные формы жирных кислот. В печени инсулин, действуя через различные механизмы, активирует ферменты путем дефосфорилирования и индуцирует их синтез. В результате увеличиваются активность и синтез ферментов, участвующих в превращении части глюкозы, поступающей с пищей в жиры. Это – регуляторные ферменты гликолиза, пируватдегидрогеназный комплекс и ферменты, участвующие в синтезе ЖК из ацетил-КоА. Результат действия инсулина на обмен углеводов и жиров в печени – увеличение синтеза жиров и секреция их в кровь в составе ЛПОНП. ЛПОНП доставляют жиры в капилляры жировой ткани, где действие ЛП-липазы обеспечивает быстрое поступление жирных кислот в адипоциты, где они депонируются в составе ТАГ.
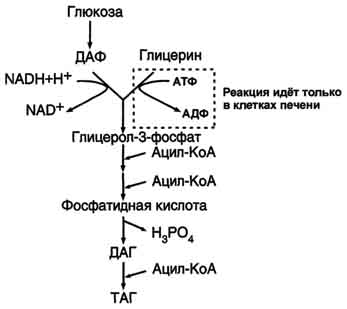
Вопрос 50. Мобилизация жира в жировой ткани (липолиз). Триацилглицерол, диацилглицерол и моноацилглицероллипазы. Гормональная регуляция липолиза в адипоцитах.
Адипоциты (место депонирования жиров) располагаются в основном под кожей, образуя подкожный жировой слой, и в брюшной полости, образуя большой и.малый сальники. Мобилизация жиров, т.е. гидролиз до глицерола и жирных кислот, происходит в постабсорбтивный период, при голодании и активной физической работе. Гидролиз внутриклеточного жира осуществляется под действием фермента гормончувствительной липазы - ТАГ-липазы. Этот фермент отщепляет одну жирную кислоту у первого углеродного атома глицерола с образованием диацилглицерола, а затем другие липазы гидролизуют его до глицерола и жирных кислот, которые поступают в кровь. Глицерол как водорастворимое вещество транспортируется кровью в свободном виде, а жирные кислоты (гидрофобные молекулы) в комплексе с белком плазмы - альбумином. Гидролиз триацилглицеролов до жирных кислот и глицерола катализируют три липазы. Первой действует триацилглицероллипаза, затем диацил-глицероллипаза и последней - моноацилглицероллипаза, гидролизующая сложноэфирную связь в моноацилглицеролах. Эти липазы локализованы в разных органоидах. Триацилглицероллипаза – в мембране липидных сферосом, моноацилглицероллипаза - в мембранах глиоксисом прорастающих семян. Указанные липазы различаются не только по специфичности, но и по оптимуму рН и температуре действия. Мобилизация депонированных жиров стимулируется глюкагоном и адреналином и, в меньшей степени, некоторыми другими гормонами (соматотропным, кортизолом). В постабсорбтивный период и при голодании глюкагон, действуя на адипоциты через аденилатциклазную систему, активирует протеинкиназу А, которая фосфо-рилирует и, таким образом, активирует гормончувствительную липазу, что инициирует липо-лиз и выделение жирных кислот и глицерина в кровь. При физической активности увеличивается секреция адреналина, который действует через β-адренергические рецепторы адипоцитов, активирующие аденилатциклазную систему (рис. 8-24). В настоящее время обнаружено 3 типа β-рецепторов: β1, β2, β3, активация которых приводит к липолитическому действию. К наибольшему липолитическому действию приводит активация β3-рецепторов. Адреналин одновременно действует и на α2-рецепторы адипоцитов, связанные с ингибирующим G-белком, что инактивирует аденилатциклазную систему. Вероятно, действие адреналина двояко: при низких концентрациях в крови преобладает его антилиполитическое действие через α2-рецепторы, а при высокой - преобладает липолитическое действие через β-рецепторы. Для мышц, сердца, почек, печени при голодании или физической работе жирные кислоты становятся важным источником энергии. Печень перерабатывает часть жирных кислот в кетоновые тела, используемые мозгом, нервной тканью и некоторыми другими тканями как источники энергии. В результате мобилизации жиров концентрация жирных кислот в крови увеличивается приблизительно в 2 раза, однако абсолютная концентрация жирных кислот в крови невелика даже в этот период. Т1/2 жирных кислот в крови тоже очень мал (менее 5 мин), что означает существование быстрого потока жирных кислот из жировой ткани к другим органам. Когда постабсорбтивный период сменяется аборбтивным, инсулин активирует специфическую фосфатазу, которая дефосфорилирует гормончувствительную липазу, и распад жиров останавливается.
Вопрос 51. Метаболизм фосфолипидов. Обмен глицерофосфолипидов. Биологическое значение различных фосфолипаз. Биосинтез фосфатидилхолина, фосфатидилэтаноламина, фосфатидилсерина.
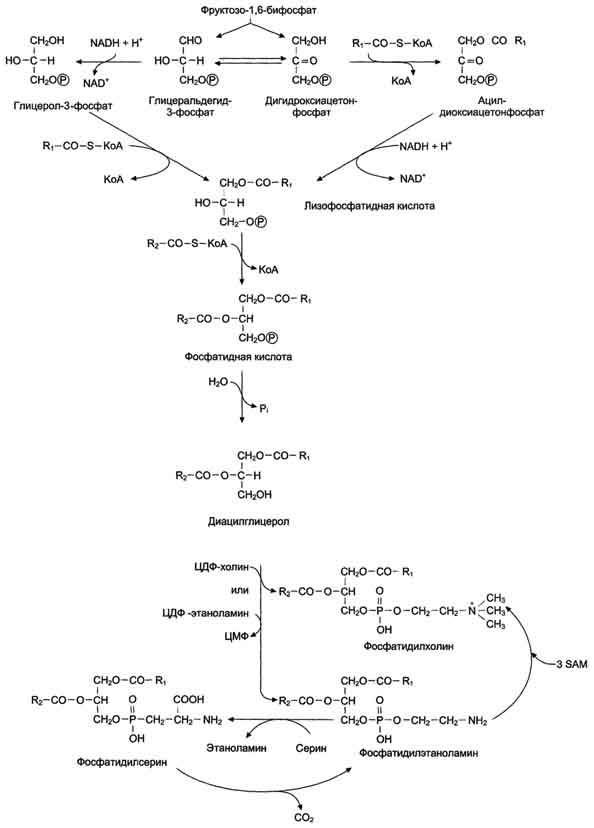
Метаболизм фосфолипидов тесно связан со многими процессами в организме: образованием и разрушением мембранных структур клеток, формированием ЛП, мицелл жёлчи, образованием в альвеолах лёгких поверхностного слоя, предотвращающего слипание альвеол во время выдоха. Нарушения обмена фосфолипидов - причина многих заболеваний, в частности, респираторного дистресс-синдрома новорождённых, жирового гепатоза, наследственных заболеваний, связанных с накоплением гликолипидов, - лизосомных болезней. При лизосомных болезнях снижается активность гидролаз, локализованных в лизосомах и участвующих в расщеплении гликолипидов. Начальные этапы синтеза глицерофосфолипидов и жиров происходят одинаково до образования фосфатидной кислоты. Фосфатидная кислота может синтезироваться двумя разными путями: через глицеральдегид-3-фосфат и через дигидроксиацетонфосфат. На следующем этапе фосфатидаза отщепляет от фосфатидной кислоты фосфатный остаток, в результате чего образуется диацилглицерол. Дальнейшие превращения диацилглицерола также могут идти разными путями. Один из вариантов - образование активной формы "полярной головки" фосфолипида: холин, серии или этаноламин превращаются в ЦДФ-холин, ЦДФ-серин или ЦДФ-этаноламин. Далее диацилглицерол взаимодействует с ЦМФ-производными, при этом выделяется ЦМФ, и образуется соответствующий фосфолипид, например фосфатидилхолин. Между глицерофосфолипидами возможны различные взаимопревращения. Фосфатидилхолин может образовываться и другим путём: из фосфатидилэтаноламина, получая последовательно 3 метальные группы от SAM. Фосфатидилсерин может превращаться в фосфа-тидилэтаноламин путём декарбоксилирования. Фосфатидилэтаноламин может превращаться в фосфатидилсерин путём обмена этаноламина на серии.
Вопрос 52. Обмен сфинголипидов. Синтез церамида и его производных. Катаболизм сфингомиелина и гликосфинголипидов, генетические дефекты энзимов.
Сфинголипиды - производные церамида, образующегося в результате соединения аминоспирта сфингозина и жирной кислоты. В группу сфинголипидов входят сфингомиелины и гликосфинголйпиды. Гликосфинголипиды - гликолипиды, в состав которых входят церамид и один или несколько остатков углеводов, и сиаловая (N-ацетилнейраминовая) кислота Гликосфинголипиды локализованы в плазматических мембранах клеток таким образом, что углеводная часть молекулы располагается на поверхности клеток и часто обладает антигенными свойствами. Синтез церамида и его производных. Синтез сфинголипидов начинается с образования церамида. Серии конденсируется с пальмитоил-КоА. Продукт их взаимодействия сначала восстанавливается коферментом NADPH, затем к аминогруппе дигидросфингозина амидной связью присоединяется жирная кислота, содержащая, как правило, 24 атома углерода. После окисления FAD-зависимой дегидрогеназой образуется церамид. Церамид служит предшественником в синтезе большой группы сфинголипидов: сфингомиелинов, не содержащих углеводов, и гликосфинголипидов Последующие реакции синтеза катализируются специфическими трансферазами, набор которых отличается в разных тканях. Соединение фосфорилхолина с церамидом сфингомиелин-синтазой приводит к образованию сфингомие-лина. Присоединение углеводных компонентов катализируется специфическими гликозилтрансферазами. Донорами углеводных компонентов служат активированные сахара: УДФ-галактоза и УДФ-глюкоза. Галактоцереброзид - главный липид миелиновых оболочек; глюкоцереброзид входит в состав мембран многих клеток и служит предшественником в синтезе более сложных гликолипидов или продуктом на пути их катаболизма.
Катаболизм сфингомиелина и его нарушения.В лизосомах находятся ферменты, способные гидролизовать любые компоненты клеток. Эти ферменты называют кислыми гидролазами,. Значение рН = 5, Катаболизм сфингомиелинов и гликолипидов происходит в лизосомах. В распаде сфингомиелинов участвуют 2 фермента - сфингомиелиназа, отщепляющая фосфорилхолин, и церамидаза, продуктами действия которой являются сфингозин и жирная кислота Генетический дефект сфингомиелиназы - причина болезни Ниманна-Пика. Дети с таким дефектом погибают в раннем возрасте. Симптомы заболевания: увеличение печени и селезёнки (гепатоспленомегалия), в лизосомах которых накапливается сфингомиелин; умственная отсталость. Генетический дефект другого фермента (церамидазы) приводит к развитию болезни Фарбера, симптомами которой также являются гепато- и спленомегалия, а также поражение суставов (болезненность, отёчность). Катаболизм гликосфинголипидов. Катаболизм гликосфинголипидов начинается с перемещения их с поверхности клетки в цитоплазму по механизму эндоцитоза. В результате молекулы, расположенные на поверхности мембран, оказываются в эндоцитозных везикулах в цитоплазме и сливаются с лизосомами. В лизосомах находятся все ферменты, необходимые для гидролиза сложных молекул гликосфинголипидов: α- и β-галактозидазы, β-глюкозидазы, нейраминидаза (сиалидаза) и церамидаза. В результате последовательных реакций гидролиза сложные молекулы гликосфинголипидов распадаются до мономеров: глюкозы, галактозы, жирной кислоты, сфингозина и других метаболитов. Генетические дефекты лизосомных ферментов катаболизма гликосфинголипидов. В норме синтез и катаболизм гликосфинголипидов сбалансированы таким образом, что количество этих компонентов в мембранах постоянно. Если имеется генетический дефект какого-либо лизосомного фермента, участвующего в катаболизме гликосфинголипида, то в лизосомах накапливается не-деполимеризованный субстрат, так называемые "остаточные тельца", размеры лизосом увеличиваются, их мембрана может разрушаться, ферменты выходят в цитозоль, и функции клеток нарушаются. Генетические заболевания вследствие дефекта какого-либо из ферментов катаболизма гликосфинголипидов называют сфинголипидоза-ми, или лизосомными болезнями. Эти заболевания редки, но среди некоторых популяций людей их частота очень высока. Так, болезнь Гоше вследствие дефекта фермента β-глюкрзидазы , болезнь Тея-Сакса (дефект фермента β-гексозаминидазы) - Сфинголипидозы обычно приводят к смерти в раннем возрасте, так как происходит поражение клеток нервной ткани, где сконцентрированы гликосфинголипиды.
Вопрос 53. Кетоновые тела. Биосинтез, использование в качестве источника энергии. Биохимический механизм кетонемии и кетонурии.
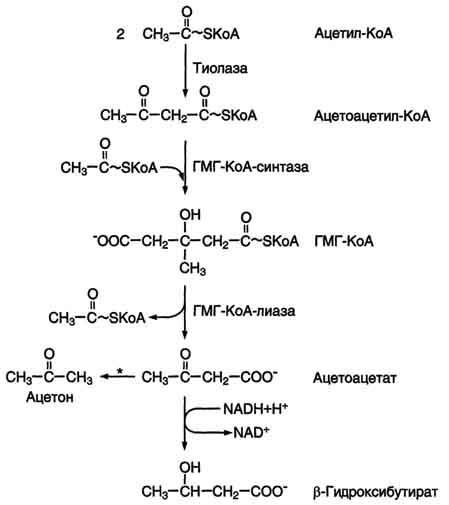
При голодании, длительной физической работе и в случаях, когда клетки не получают достаточного количества глюкозы, жирные кислоты. используются многими тканями как основной источник энергии. В отличие от других тканей мозг и другие отделы нервной ткани практически не используют жирные кислоты в качестве источника энергии. В печени часть жирных кислот превращается в кетоновые тела, которые окисляются мозгом, нервной тканью, мышцами, обеспечивая достаточное количество энергии для синтеза АТФ и уменьшая потребление глюкозы. К кетоновым телам относят β-гидроксибутират, ацетоацетат и ацетон. Первые две молекулы могут окисляться в тканях, обеспечивая синтез АТФ. Ацетон образуется только при высоких концентрациях кетоновых тел в крови и, выделяясь с мочой, выдыхаемым воздухом и потом, позволяет организму избавляться от избытка кетоновых тел. Синтез кетоновых тел в печени. При низком соотношении инсулин/глюкагон в крови в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, поэтому увеличивается скорость β-окисления (рис. 8-32). Скорость реакций ЦТК в этих условиях снижена, так как оксалоацетат используется для глюконеогенеза. В результате скорость образования ацетил-КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза кетоновых тел. Синтез кетоновых тел происходит только в митохондриях печени.
Синтез кетоновых тел начинается с взаимодействия двух молекул ацетил-КоА, которые под действием фермента тиолазы образуют ацетоацетил-КоА (рис. 8-33). С ацетоацетил-КоА взаимодействует третья молекула ацетил-КоА, образуя 3-гидрокси-3-метилглутарил-КоА (ГМГ-КоА). Эту реакцию катализирует фермент ГМГ-КоА-синтаза. Далее ГМГ-КоА-лиаза катализирует расщепление ГМГ-КоА на свободный ацетоацетат и ацетил-КоА. Ацетоацетат может выделяться в кровь или превращаться в печени в другое кетоновое тело - β-гидроксибутират путём восстановления. В клетках печени при активном β-окислении создаётся высокая концентрация NADH. Это способствует превращению большей части ацетоацетата в β-гидроксибутират, поэтому основное кетоновое тело в крови - именно β-гидроксибутират. При голодании для многих тканей жирные кислоты и кетоновые тела становятся основными топливными молекулами. Глюкоза используется в первую очередь нервной тканью и эритроцитами. При высокой концентрации ацетоацетата часть его неферментативно декарбоксилируется, превращаясь в ацетон. Ацетон не утилизируется тканями, но выделяется с выдыхаемым воздухом и мочой. Таким путём организм удаляет избыточное количество кетоновых тел, которые не успевают окисляться, но, являясь водорастворимыми кислотами, вызывают ацидоз.Кетоацидоз. В норме концентрация кетоновых тел в крови составляет 1-3 мг/дл (до 0,2 мМ/л), но при голодании значительно увеличивается. Увеличение концентрации кетоновых тел в крови называют кетонемией, выделение кетоновых тел с мочой - кетонурией. Накопление кетоновых тел в организме приводит к кетоацидозу: уменьшению щелочного резерва (компенсированному ацидозу), а в тяжёлых случаях - к сдвигу рН (некомпенсированному ацидозу), так как кетоновые тела (кроме ацетона) являются водорастворимыми органическими кислотами (рК
3,5), способными к диссоциации:
СН3-СО-СН2-СООН ↔ СН3-СО-СН2-СОО- + Н+.
Ацидоз достигает опасных величин при сахарном диабете, так как концентрация кетоновых тел при этом заболевании может доходить до 400-500 мг/дл. Тяжёлая форма ацидоза - одна из основных причин смерти при сахарном диабете. Накопление протонов в крови нарушает связывание кислорода гемоглобином, влияет на ионизацию функциональных групп белков, нарушая их кон-формацию и функцию.
|
|
|
 Скачать 2.83 Mb.
Скачать 2.83 Mb.