конспект лекций.Химия(органическая химия).dos. конспект лекций.Химия(органическая химия). Конспект лекций по дисциплине для студентов, обучающихся по специальностям и направлениям 050100. 62Естественнонаучное образование
 Скачать 1.49 Mb. Скачать 1.49 Mb.
|

|
б) Геометрическая изомерия Геометрические изомеры возникают в результате отсутствия в молекуле: 1. вращения атомов углерода относительно друг друга - следствие жесткости двойной связи С=С или циклической структуры; 2. двух одинаковых групп при одном атоме углерода двойной связи или цикла. Геометрические изомеры, в отличие от конформеров, могут быть выделены в чистом виде и существуют как индивидуальные, устойчивые вещества. Для их взаимного превращения необходима более высокая энергия - порядка 125-170 кДж/моль (30-40 ккал/моль). Различают цис-транс-(Z,E) изомеры; цис-формами называют геометрические изомеры, у которых одинаковые заместители лежат по одну сторону от плоскости π-связи или цикла, транс-формами называют геометрические изомеры, у которых одинаковые заместители лежат по разные стороны от плоскости π-связи или цикла. Простейшим примером могут служить изомеры бутена-2, который существует в виде цис- , транс-геометрических изомеров: цис-бутен-2 транс-бутен-2 температура плавления -138,90С - 105,60С температура кипения 3,720С 1,000С плотность 0,724 0,604 1,2 – дихлорциклопропан существует в виде цис- , транс-изомеров: 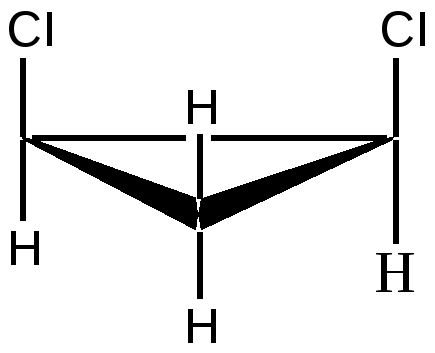 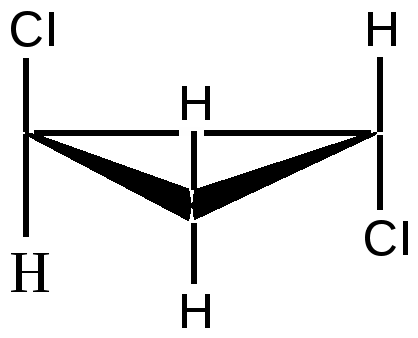 цис-1,2-дихлорциклопропан транс-1,2-дихлорциклопропан В более сложных случаях применяется Z,E-номенклатура (номенклатура Канна, Ингольда, Прелога – КИП, номенклатура старшинства заместителей). В соединении 1-бром -2-метил-1-хлорбутене-1 (Br)(CI)С=С(СН3) - СН2-СН3 все заместители при атомах углерода с двойной связью различные; поэтому данное соединение существует в виде Z-, E- геометрических изомеров: 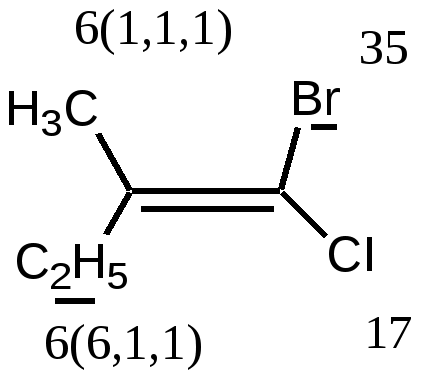  Е-1-бром-2-метил-1-хлорбутен-1 Z-1-бром-2-метил-1-хлорбутен-1. Для обозначения конфигурации изомера указываютрасположение старших заместителей при двойной связи (или цикле) – Z-(от немецкого Zusammen - вместе) или Е-(от немецкого Entgegen - напротив). В Z,E-системе старшими считаются заместители с большим порядковым (атомным) номером. Если атомы, непосредственно связанные с ненасыщенными атомами углерода, одинаковы, то переходят ко "второму слою", в случае необходимости - к "третьему слою" и т. д. В первой проекции старшие группы находятся напротив друг друга относительно двойной связи, поэтому это Е-изомер. Во второй проекции старшие группы расположены по одну сторону относительно двойной связи (вместе), поэтому это Z-изомер. Геометрические изомеры широко распространены в природе. Например, природные полимеры каучук (цис-изомер) и гуттаперча (транс-изомер), природная фумаровая (транс-бутендиовая кислота) и синтетическая малеиновая (цис-бутендиовая кислота) кислоты, в составе жиров - цис-олеиновая, линолевая, линоленовая кислоты. в) Оптическая изомерия Молекулы органических соединений могут быть хиральными и ахиральными. Хиральность(от греч. сheir - рука) — несовместимость молекулы со своим зеркальным отражением. Хиральные вещества способны вращать плоскость поляризации света. Это явление называют оптической активностью, а соответствующие вещества - оптически активными. Оптически активные вещества встречаются в виде пар оптических антиподов - изомеров, физические и химические свойства которых в обычных условиях одинаковы, за исключением одного - знака вращения плоскости поляризации: один из оптических антиподов отклоняет плоскость поляризации в право (+, правовращающий изомер), другой – влево (-, левовращающий). Определить конфигурацию оптических антиподов можно экспериментально с помощью прибора - поляриметра. Оптическая изомерия появляется тогда, когда в молекуле присутствует асимметрический атом углерода (существуют и другие причины хиральности молекулы). Так называют атом углерода в sр3 - гибридизации и связанный с четырьмя различными заместителями. Возможны два тетраэдрических расположения заместителей вокруг асимметрического атома. При этом две пространственные формы нельзя совместить никаким вращением; одна из них является зеркальным изображением другой : 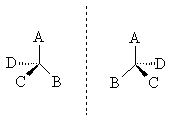 Обе зеркальные формы составляют пару оптических антиподов или энантиомеров. Изображают оптические изомеры в виде проекционных формул Э. Фишера. Их получают в результате проецирования молекулы с асимметрическим атомом углерода. При этом сам асимметрический атом углерода на плоскости обозначают точкой, на горизонтальной линии указывают символы заместителей, выступающих перед плоскостью рисунка. На вертикальной линии (прерывистой или сплошной) указывают заместители, которые удалены за плоскость рисунка. Ниже приведены различные способы записи проекционной формулы, отвечающей левой модели на предыдущем рисунке:  В проекции главную углеродную цепь изображают вертикально; главную функцию, если она находится в конце цепи, указывают в верхней части проекции. Например, стереохимические и проекционные формулы (+) и (-) аланина - СН3 -*СН(NН2)-СООН представляют следующим образом:  Смесь с одинаковым содержанием энантиомеров называется рацематом. Рацемат не обладает оптической активностью и характеризуется отличными от энантиомеров физическими свойствами. Правила преобразования проекционных формул. 1. Формулы можно вращать в плоскости чертежа на 180о, не меняя их стереохимического смысла: 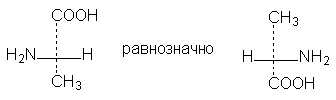 2. Две (или любое четное число) перестановки заместителей у одного асимметрического атома не меняют стереохимического смысла формулы: 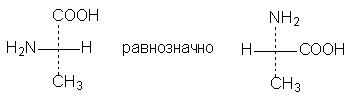 3. Одна (или любое нечетное число) перестановка заместителей у асимметрического центра приводит к формуле оптического антипода:  4. Поворот в плоскости чертежа на 90о превращает формулу в антипод. 5. Вращение любых трех заместителей по часовой стрелке или против не меняет стереохимического смысла формулы: 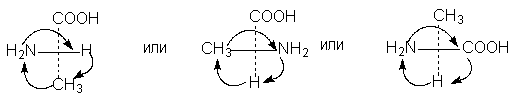 6. Проекционные формулы нельзя выводить из плоскости чертежа. Оптической активностью обладают органические соединения, в молекулах которых хиральными центрами являются и другие атомы, например кремния, фосфора, азота, серы. Соединения с несколькими асимметрическими атомами углерода существуют в виде диастереомеров, т.е. пространственных изомеров, не составляющих друг с другом оптических антиподов. Диастереомеры отличаются друг от друга не только оптическим вращением, но и всеми другими физическими константами: у них разные температуры плавления и кипения, разные растворимости и др. Число пространственных изомеров определяют по формуле Фишера N=2n, где n - число асимметрических атомов углерода. Число стереоизомеров может уменьшаться из-за частичной симметрии, появляющейся в некоторых структурах. Оптически неактивные диастереомеры называют мезо-формами. Номенклатура оптических изомеров: а) D- , L- номенклатура Для определения D- или L-ряда изомера конфигурацию (положение ОН–группы у асимметричного атома углерода) сравнивают с конфигурациями энантиомеров глицеринового альдегида (глицериновый ключ):  L-глицериновый альдегид D-глицериновый альдегид Применение D-, L-номенклатуры в настоящее время ограничено тремя классами оптически активных веществ: углеводами, аминокислотами и оксикислотами. б) R -, S-номенклатура (номенклатура Кана, Ингольда и Прелога) Для определения R(правый)- или S(левый)-конфигурации оптического изомера необходимо расположить заместители в тетраэдре (стереохимической формуле) вокруг асимметрического углеродного атома таким образом, чтобы самый младший заместитель (обычно это водород) имел направление «от наблюдателя». Если переход трех остальных заместителей от старшего к среднему и младшему по старшинству происходит по часовой стрелке - это R-изомер (падение старшинства совпадает с движением руки при написании верхней части буквы R). Если переход происходит против часовой стрелки - это S-изомер (падение старшинства совпадает с движением руки при написании верхней части буквы S). Для определения R- или S-конфигурации оптического изомера по проекционной формуле необходимо путем четного числа перестановок расположить заместители так, чтобы самый младший из них оказался внизу проекции. Падение старшинства остальных трех заместителей по часовой стрелке соответствует R-конфигурации, против часовой стрелки - S-конфигурации. Получают оптические изомеры следующими методами: а) выделение из природных материалов, содержащих оптически активные соединения, например белки и аминокислоты, углеводы, многие оксикислоты (винная, яблочная, миндальная), терпеновые углеводороды, терпеновые спирты и кетоны, стероиды, алкалоиды и др. б) расщепление рацематов; в) асимметрический синтез; г) биохимическое получение оптически активных веществ. ЗНАЕТЕ ЛИ ВЫ, ЧТО -Явление изомерии (от греч.- isos— разный и meros- доля, часть) открыто в 1823г. Ю. Либихом и Ф. Вёлером на примере солей двух неорганических кислот: циановой Н-О-С≡N и гремучей Н-О-N= С. -В 1830 г. Ж.Дюма распространил представление об изомерии на органические соединения. -В 1831г. термин «изомер» для органических соединений предложил Й. Берцелиус. -Стереоизомеры природных соединений характеризуются разной биологической активностью (аминокислоты, углеводы, алкалоиды, гормоны, феромоны, лекарственные вещества природного происхождения и т.д.). ЛЕКЦИЯ № 5. КЛАССИФИКАЦИЯ ОРГАНИЧЕСКИХ РЕАКЦИЙ И РЕАГЕНТОВ. ОРГАНИЧЕСКИЕ КИСЛОТЫ И ОСНОВАНИЯ План 1. Классификация органических реакций: а) по характеру изменения связей в реагирующих веществах; б) по конечному результату (или по направлению) реакции. 2. Кислотно-основные взаимодействия. Органических реакций очень много, однако, используя различные критерии, их можно классифицировать. В результате всё многообразие реакций можно свести к небольшому числу типов реакций. Взаимодействующие в органической реакции вещества подразделяют на реагент и субстрат. При этом считается, что реагент атакует субстрат. Субстратом, как правило, считают молекулу, которая предоставляет атом углерода для новой связи. Классификация органических реакций: 1. по характеру изменения связей в реагирующих веществах реакции подразделяют на радикальные и ионные. а) Радикальные реакции протекают с участием радикалов (R.) - частиц с неспаренным электроном и образуемых в результате гомолитического разрыва ковалентной связи, например CI:CI → ·CI + ·CI. Для радикалов характерна высокая реакционная активность, реакции с их участием протекают с очень большой скоростью. Примеры радикальных реагентов: ·CI,·Br, ·J, ·NO2, ·OH, ·R(алкил) и др. б) Ионные реакции протекают с участием ионов, образуемых в результате гетеролитического разрыва ковалентной связи: Е :N → Е+ + :N-. Электрофилы (электро+фил - любящий электрон) (Е): Br+, Cl+, H+, R+, NO2 + и нейтральные молекулы с электронодефицитным центром - SO3, BF3, соли алюминия, цинка, железа (III) и др. Электрофил представляет незаполненные, вакантные орбитали для образования ковалентной связи. Нуклеофилы (нуклеос+фил - любящий протон) (N): Hal-, OH-, RO-, RS-, RCOO-, R-, CN- и нейтральные молекулы с неподеленной электронной парой, например H2O:, ROH, :NH3, RNH2 и др.За счет пары электронов нуклеофил способен образовывать новую ковалентную связь. 2. по конечному результату (или по направлению) реакции подразделяют: а) реакции присоединения - символ А (анг.-addition).Присоединение реагента к субстрату происходит по π-связям или по σ-связям циклических структур (размыкание цикла), в результате реакций образуются новые ковалентные σ-связи. Реакции присоединения могут быть электрофильными (АЕ): нуклеофильными (АN): р  . Реакции присоединения водорода называют гидрированием, воды - гидратацией, галогенов - галогенированием (хлорирование, бромирование и т.д.), галогеноводородов - гидрогалогенированием и др. б) реакции замещения - символ S (анг.-substitution).Замещение происходит по σ-связям субстрата, в результате реакций образуются новые ковалентные σ-связи. Реакции замещения могут быть электрофильными (SЕ): нуклеофильными (SN): Н3Сδ+-Оδ- H + Нδ+-Clδ- → Н3С-Cl + HОН, радикальными (SR): Н3С-H + Cl-Cl → Н3С-Cl + HCl. в) реакции отщепления или элиминирования - символ Е (анг.- elimiation). Отщепление происходит по σ-связям субстрата. В результате α, ß-отщепления образуются новые π-связи, в результате α,γ- или α,δ-отщепления образуются новые ковалентные σ-связи циклических соединений. Например: СI-СН2-СН2-СН2-СI + Zn→ + Zn СI2 Реакции отщепления водорода называют дегидрированием, воды - дегидратацией, галогенов - дегалогенированием (дехлорирование, дебромирование и т.д.), галогеноводородов - дегидрогалогенированием и др. г) перегруппировки. В процессе перегруппировок внутри молекулы происходит миграция атома или групп атомов от одного атома к другому. Например: д) окислительно-восстановительные. Окислительно-восстановительный характер органических реакций определяют иначе по сравнению с неорганическими реакциями. Так, окисление - образование новых связей углерода с атомами более электроотрицательных элементов (галогены, кислород, азот, сера и т.д.), иногда в реакциях этого типа число атомов водорода в продукте реакции может уменьшаться. Например: СН3-СН2-ОН + [О] → СН3-СН=О Восстановление - образование новых связей С-Н, при этом часто число атомов водорода в продукте реакции увеличивается. Например: СН3-СН=СН-СН3 + Н2 → СН3-СН2-СН2-СН3 е) по числу реагирующих частиц. Большинство органических реакций состоят из нескольких последовательных или параллельных элементарных стадий. В зависимости от числа частиц, участвующих в скорость-определяющей стадии (самой медленной или лимитирующей), различают мономолекулярные и бимолекулярные реакции. Например, реакции мономолекулярного и бимолекулярного нуклеофильного замещения (символы SN1 и SN2), мономолекулярного и бимолекулярного отщепления (символы Е1 и Е2) и др. Кислотно-основные взаимодействия В настоящее время существуют две основных теории кислот и оснований: теория Брёнстеда-Лоури (1923 г.) и теория Льюиса (1926 г.). Кислоты Брёнстеда – это соединения, способные отдавать протон (доноры протона). Основания Брёнстеда – это соединения, способные присоединять протон (акцепторы протона). Для взаимодействия с протоном основание должно иметь свободную пару электронов или электроны π-связи. Кислоты и основания образуют сопряженные кислотно-основные пары: В зависимости от природы элемента, с которым связан протон, различают четыре основных типа органических кислот Брёнстеда: O-H-кислоты - карбоновые кислоты, спирты, фенолы; S-H-кислоты - тиолы; N-H-кислоты - амины, амиды, имиды; C-H-кислоты - углеводороды и их производные. Мерой силы кислоты является константа кислотности (ионизации) Ка или рКа. Чем больше Ка (или меньше рКа), тем сильнее кислота. В зависимости от природы атома, к неподеленной паре электронов которого присоединяется протон, основания Брёнстеда делят на три основных типа: N (аммониевые) основания - амины, нитрилы, азотсодержащие гетероциклические соединения; О (оксониевые) основания - спирты, простые эфиры, альдегиды, кетоны, карбоновые кислоты и их функциональные производные; S (сульфониевые) основания - тиолы, сульфиды. Особый тип оснований Бренстеда представляют π-основания, в которых центром основности являются электроны π-связи (алкены, арены). При прочих равных условиях для элементов одного периода с ростом электроотрицательности атома кислотность соединений увеличивается, так как высокая электроотрицательность атома при кислотном центре стабилизирует образующийся при отщеплении протона анион. Так, кислотность уменьшается в ряду: OH-кислоты> NH-кислоты> CH-кислоты Для элементов одной подгруппы с возрастанием заряда ядра и поляризуемости атома кислотность соединений увеличивается: OH-кислоты < SH-кислоты Введение заместителя в углеводородный радикал влияет на силу кислоты. Электроноакцепторные (ЭА) заместители увеличивают, а электронодонорные (ЭД) - уменьшают кислотность, поскольку электроноакцепторные заместители стабилизируют сопряженное основание (анион), а электронодонорные заместители - дестабилизируют. Основность уменьшается в ряду: N-основания > О-основания > S-основания (NOS) Введение электронодонорных заместителей увеличивает, а введение электроакцепторных - понижает основность. Дж. Льюисом была предложена более общая теория кислот и оснований. Основания Льюиса – это доноры пары электронов (спирты, алкоголят-анионы, простые эфиры, амины и т.д.) Кислоты Льюиса – это акцепторы пары электронов, т.е. соединения, имеющие вакантную орбиталь (ион водорода и катионы металлов: H+, Ag+, Na+, Fe2+; галогениды элементов второго и третьего периодов BF3, AlCl3, FeCl3, ZnCl2; галогены; соединения олова и серы: SnCl4, SO3). Кислотно-основное взаимодействие по Льюису - это доноро-акцепторное взаимодействие и любую гетеролитическую реакцию можно представить как взаимодействие кислоты и основания Льюиса. |