Физ химия курс лекций. Лекция 1 Тема Химическая термодинамика
 Скачать 2.63 Mb. Скачать 2.63 Mb.
|

Рис. 3.3. Диаграмма потенциальной энергии дляединичного столкновения реагентов Р: АК – активированный комплекс; П - продукты Активированный комплекс (АК) для рассмотренной реакции можно представить следующим образом: [Н3С···Н···Сl ]. В общем виде взаимодействие молекул А2 и В2 будет протекать по уравнению:  реагенты активированный продукты (начальное состояние) комплекс (конечное состояние) Разность потенциальной энергии активированного комплекса и реагентов называют энергией активации ЕА. Если в реакции (3.9) будет участвовать по одному молю частиц реагентов, то стандартным условиям будет соответствовать диаграмма, представленная на рис. 3.4а, которую называют энтальпийным или энергетическим профилем реакции. Она показывает соотношение между а) б) 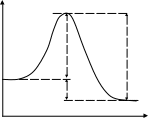 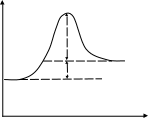 AK АК AK АК  Еа Р r r Рис. 3.4. Энтальпийные профили реакцийстандартными энтальпиями 1 моля реагентов, активированных комплексов и продуктов. ЛЕКЦИЯ 13 3.4. СЛОЖНЫЕ ХИМИЧЕСКИЕ РЕАКЦИИ В основе кинетики сложных химических реакций лежит принцип независимости реакций, согласно которому: если в системе протекает несколько реакций, то каждая из них протекает независимо от других, так что общее изменение в системе равно сумме этих независимых изменений. В последовательных реакциях продукт одной реакции является исходным веществом для другой реакции. Рассмотрим схему простейшего примера: А = В = С, в котором вещество В является промежуточным. Согласно кинетическим кривым рис.3.5, концентрация исходного вещества снижается, а продукта - увеличивается. Концентрация В вначале увеличивается, достигая максимума, а затем уменьшается, так как расход вещества В не компенсируется его образованием из вещества А. 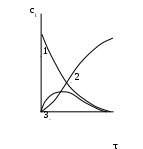 Рис. 3.5. Зависимость концентрации веществ от времени (кинетические кривые) при протекании сложной реакции, состоящей из двух последовательных стадий: 1 – исходное вещество; 2 – продукт реакции; 3 – промежуточное вещество Если константы скоростей отдельных стадий близки по значениям, то константа скорости суммарной реакции будет равна их величине: k1 ≈ k2 ≈ ki ≈ ≈ k. Если же константы скоростей отдельных стадий отличаются друг от друга значительно, то скорость суммарной реакции будет определяться самой медленно идущей стадией: k = kmin. Примером последовательно идущих реакций может служить процесс образования оксида азота IV из азота и кислорода воздуха в условиях работы двигателей внутреннего сгорания (автомобилей). Вначале при высокой температуре горения топлива образуется оксид азотаII: N2 + O2 = 2NO, который затем частично окисляется кислородом до оксида азотаIV: 2NO + O2 = 2NO2 . В параллельныхреакциях, например, исходное вещество одновременно расходуется в двух различных реакциях:  k1 В k1 В  А АM k2 Тогда скорость суммарной реакции по отношению к веществу А будет складываться по двум скоростям: vA = - dc(А)/d = (k1 + k2) c(А). Скорости получения продуктов: dc(В)/d = k1c(А) ; dc(М)/d = k2c(А). После интегрирования и преобразования получим, что концентрации продуктов пропорциональны константам скоростей: c(В)/c(М) = k1 / k2 . Полученное соотношение используется для изучения кинетики быстрых реакций. Если образование вещества В происходит очень быстро, а кинетику образования М можно изучить в ходе независимых экспериментов, то по соотношению концентраций продуктов можно установить кинетические параметры первой конкурирующей реакции. На рис. 3.6 показано изменение концентраций веществ для случая k1 k2.  с с  c(В) c(М) c(В) c(М) c(А)  τ τРис. 3.6. Зависимость концентраций реагента (А) и продуктов (В и М) в реакционной системе от времени Низкая селективность реакций характерна для органических веществ. Например, при хлорировании метана одновременно получаются CH3Cl, CH2Cl2, CHCl3, CCl4. При нитровании фенола образуются орто-нитрофенол и пара-нитрофенол. Сопряженными называются такие реакции, которые не могут протекать друг без друга, то есть когда одна самопроизвольная реакция вызывает протекание другой реакции. Например, хромовая кислота не окисляет иодоводород в отсутствии оксида железаII:   H2CrO4 + 2FeO H2CrO4 + 2FeO Cr2O3 + Fe2O3 + 4H2O + 2I2 . H2CrO4 + 4HI Cr2O3 + Fe2O3 + 4H2O + 2I2 . H2CrO4 + 4HI Коррозия металла невозможна без наличия окислителя в окружающей среде:  2Zn - 4e 4Zn(OH)2 . 2Zn - 4e 4Zn(OH)2 . O2 + 2H2O + 4e O2 + 2H2O + 4eПринцип независимости реакций неприменим к сопряженным реакциям. В цепных реакциях на каждом этапе ее протекания возникает активная частица (радикал), которая вызывает цепь дальнейших превращений. Активными являются частицы с нескомпенсированной валентностью, то есть имеющие неспаренные электроны: Каждая цепная реакция включает три основные стадии: зарождение цепи, продолжение, обрыв. Зарождение цепи обычно начинается с разрыва наименее прочной молекулы, который происходит с низкой энергией активации. Первая стадия чаще всего осуществляется в результате инициирования извне: под действием высокой температуры, излучения, добавления специальных веществ. Например, при облучении газообразного хлора синим светом происходит диссоциация его молекул на атомы: Химическими инициаторами могут выступать вещества, которые легко распадаются на свободные радикалы, например, органические перекиси и хлористый нитрозил: Стадия продолжения цепи состоит из большого числа повторяющихся элементарных реакций взаимодействия активных частиц с исходными веществами. Так, схеме неразветвленной цепи соответствует процесс хлорирования этилена: Cl2 + C2H4 = C2H4Cl2 ; Видно, что радикал хлора воспроизводится, и процесс повторяется множество раз. Схема разветвленной цепи предусматривает увеличение числа активных частиц на отдельных стадиях:      и так далее. Примером такой реакции является горение водорода в кислороде. В простейшем виде реакцию можно представить в виде схемы: Цепь I Цепь II 1. 2. 1. 2. Цепь I состоит из чередующихся стадий 1, 2, 1, 2 …, цепь II – из стадий 3, 2, 1, 3, 2, 1 …. При этом только в одной стадии (второй) образуется молекула продукта (вода) и одна активная частица ( Обрыв цепной реакции происходит в результате двойного столкновения радикалов со стенкой сосуда или в результате тройного столкновения двух радикалов с некоторой частицей М, которая забирает на себя избыточную энергию, выделяющуюся при образовании химической связи. При двойном столкновении радикалов между собой цепь не обрывается, так как образовавшаяся молекула имеет большой запас энергии, является неустойчивой и вновь распадается на радикалы. В роли частиц М могут выступать молекулы, имеющие прочные химические связи, и атомы благородных газов, например: N2, CO, Ar, Kr и т.д.. При этом молекулы М приобретают возбужденное состояние М*, но их распада не происходит. Существует множество реакций с нетермической активацией реагирующих частиц. Реакции, протекающие под воздействием быстрых элементарных частиц, являются предметом изучения радиационной химии, а протекающие в плазме электрического разряда - плазмохимии. Если реакция происходит при поглощении квантов электромагнитного излучения, в том числе квантов света, то реакция называется фотохимической. Характерными особенностями реакций с нетермической активацией являются: независимость скорости реакций от температуры; возникновение значительно большего количества активных частиц по сравнению с реакциями с термической активацией. Понятие свет включает видимый спектр излучения и ультрафиолетовую область. Первый закон фотохимии Гротгуса-Дрепера гласит: только поглощаемое средой световое излучение может произвести ее химическое изменение. Отсюда следует, что вещества, реагирующие под воздействием видимого спектра излучения, окрашены (хлорофилл, хлор, оксид азота NO2 и т.д.). Многие системы, однако, поглощают световую энергию без каких-либо химических изменений. Второй закон фотохимии сформулировал Эйнштейн: каждый поглощенный квант света в первичном акте способен активировать только одну молекулу. Отсюда следует, что 1 моль вещества поглощает число Авогадро квантов: Е(М) = NAh = NAhс/ , где - частота излучения; h - постоянная Планка; - длина волны излучения; с - скорость света. Чем выше частота (чем меньше длина волны), тем химически активнее излучение. В области видимого спектра наименьшей химической активностью обладают красные, а наибольшей - фиолетовые лучи. При поглощении кванта света молекула переходит в возбужденное состояние: М + h = М*. Энергия электромагнитного излучения трансформируется в энергию движения электронов примерно за 10-15 с. После этого могут происходить следующие превращения: переход в изомерную молекулу, например: триплет синглет 1 синглет 2; распад на радикалы Cl2* = 2 реакция с другими молекулами в системе; ионизация Основной количественной характеристикой фотохимической реакции является квантовый выход , который равен следующему соотношению: Согласно закону Эйнштейна квантовый выход в истинно фотохимической реакции равен 1. Однако на самом деле может быть и больше, и меньше 1. Если 1, то после поглощения кванта молекула может не успеть прореагировать и отдать избыточную энергию другой молекуле при столкновении; либо молекула может испустить квант света (свечение вещества вследствие испускания энергии возбуждения называется люминесценцией). Квантовый выход может составить несколько единиц, так как вслед за фотохимической реакцией могут идти темновые реакции без участия квантов света. Например, в стратосфере существует озоновый слой, который защищает Землю от губительного ультрафиолетового излучения, и в этом слое протекают следующие реакции: 1) О2 + h (дальний ультрафиолет) = 2О - фотохимическая стадия; 2О2 + 2О + М = 2О3 + М* - темновая стадия. 3О2 + h = 2О3 2) О3 + h (ближний ультрафиолет) = О2 + О - фотохимическая стадия; О + О3 = 2О2 - темновая стадия. 2О3 + h = 3О2 В обоих случаях на две молекулы озона приходится один квант света (). Если 1 и достигает 106, то протекает цепная реакция. Скорость фотохимической реакции пропорциональна молярной концентрации вещества, поглощающего свет: v = kc. 3.5. КАТАЛИЗ Катализатором называют вещество, увеличивающее скорость химической реакции, но не входящее в состав конечных продуктов. Ингибитором называют вещество, замедляющее скорость реакции. Поскольку G реакции не зависит от пути процесса, в том числе от участия катализатора, константа равновесия при этом не изменяется. Таким образом, катализатор не смещает химическое равновесие, а лишь ускоряет его достижение. Если реакция термодинамически запрещена (G), то и в присутствии катализатора она не может быть осуществлена. Различают гомогенный и гетерогенный катализ. При гомогенном катализе вещества и катализатор находятся в одной фазе. Гомогенно-каталитические реакции в газовой фазе обычно протекают по механизму цепных реакций. В качестве примера приведем механизмы разрушения озонового слоя в присутствии оксидов азота (они образуются из компонентов воздуха при полетах сверхзвуковых самолетов и космических аппаратов) и фреонов (они используются в кондиционерах и холодильных установках). I) NO + O3 = NO2 + O2 II) NO2 + O = NO + O2 O3 + O = 2O2 , O3 + O = 2O2 . В первом случае катализатором является оксид азотаII, а во втором - радикалы хлора. При этом каждая частица катализатора может разрушить тысячи молекул озона, в результате таких процессов появляются так называемые озоновые дыры. При гетерогенном катализе катализатор находится в иной фазе, чем реагенты, и реакция протекает на границе раздела фаз (например, окисление аммиака на поверхности платины в производстве азотной кислоты). Было установлено, что катализатор образует промежуточные продукты с реагентами в промежуточных реакциях (стадиях), протекающих c более низкими значениями энергии активации по сравнению с энергией активации реакции без катализатора. Именно поэтому скорость суммарного процесса в присутствии катализатора выше скорости реакции без катализатора. Взаимодействие катализатора с реагентами может протекать по слитному или по раздельному механизмам. При слитном процессе в состав активированных комплексов входят все вещества, включая катализатор К: А + В → [А…В] → АВ - без катализатора; А + В + К = [А…В…К] → АВ + К - с катализатором. По слитному механизму протекают многие реакции гомогенного и ферментативного катализа в области температур 300 - 400 К. Например, кислоты действуют каталитически на многие вещества: R 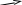 -CO - OR` + H+ = R-CO-O+R` -CO - OR` + H+ = R-CO-O+R` | Н активированный комплекс При раздельном механизме процесс участия катализатора может включать несколько стадий: А + К →[А…К] → АК АК + В → [А…К…В] → АВ + К. Реакции с раздельным механизмом происходят главным образом гетеролитически при 600 - 800 К. Если реакция может протекать по нескольким различным путям, то замена катализатора может вызвать преимущественное протекание ее по другом определенному пути. В этом проявляется селективность катализатора. Например, диэтиловый эфир без катализатора разлагается преимущественно по реакции: С2Н5ОС2Н5 = 2СН4 + 1/2С2Н4 + СО, а в присутствии йода: С2Н5ОС2Н5 = С2Н6 + СН3СНО. Специфичность каталитического действия заключается в том, что реакции данного типа ускоряются катализаторами определенного химического состава, независимо от того, являются они гомогенными или гетерогенными. Так, кислотно-основные реакции ускоряются кислотами или основаниями, а окислительно-восстановительные процессы - переходными металлами и их соединениями (особенно сильно - металлами восьмой группы). Специфичность действия особенно ярко проявляется у ферментов - биологических катализаторов, число которых в организмах составляет десятки тысяч. Часто в образовании химической связи с реагентами участвуют определенные группы атомов катализатора, которые называют активными центрами катализатора. Если активные центры блокируются нежелательными примесями в системе (каталитическими ядами), то катализатор теряет свою активность по отношению к данной реакции. Каталитическими ядами для многих катализаторов являются соединения серы, мышьяка, ртути. Некоторые другие примеси, называемые промоторами, сами по себе не проявляют каталитической активности, но могут изменять строение активных центров катализатора и усиливать их действие. Каталитическая активность Ак катализатора показывает увеличение скорости реакции vк по сравнению со скоростью реакции без катализатора vо: Ак = vк - vо(1), где - доля объема системы, занимаемая катализатором. Для сравнения каталитической активности различных катализаторов каталитическую активность можно отнести к единице массы катализатора, к его объему, к единице поверхности для гетерогенного катализатора, к концентрации в случае гомогенного катализа. В случае ингибирования процесса катализатор как бы заманивает реакцию в энергетическую западню. Реагенты и катализатор образуют нестабильный продукт по реакции с меньшей энергией активации, но распад этого продукта на новые вещества сопровождается очень высокой энергией активации, и поэтому реакционная система возвращается в исходное состояние. ЛЕКЦИЯ 14 |