Струков Патологическая анатомия. Литература для студентов медицинских институтов А. И. Струков В. В. Серов Патологическая анатомия Издание второе
 Скачать 16.97 Mb. Скачать 16.97 Mb.
|

|
Рис. 371. Схематическое изображение терато- генного терминационного периода отдельных органов и частей тела (по Гертлеру). Известно, что при поражении эмбриона виру- сом краснухи возникает рубеолярная эмбриопа- тия (синдром Грегга), которая заключается в по- роках развития глаз, сердца, мозга, зубных за- чатков и внутреннего уха. При этом пороки раз- вития глаз (катаракта, микрофтальмия и др.) по- являются в тех случаях, если мать переносит крас- нуху в последнюю декаду I месяца или в первые две декады II месяца беременности, пороки развития мозга (микроцефалия) — в тече- ние всего II месяца, внутреннего уха — в тре- тью декаду II и в первую декаду III месяца беременности. Для каждого органа существует определенный отрезок времени, в течение которого при воздействии тератогенного агента возникает порок развития этого органа. Этот отрезок времени получил название тератогенного терминационного периода (от лат. teratos — уродство и terminus — предел, граница), т. е. предельного срока, в течение которого тератогенный фактор может вызывать врожденный порок (рис. 371). Пользуясь данными эмбриологии, можно судить о сроках возникновения того или иного порока развития и составить так называемые тератологические календари для поро- ков развития различных органов. Как показывают данные экспериментальной тератологии, чувствительность развивающегося органа к повреждающим агентам определяется степенью его митотической активности: чем выше ми- тотическая активность развивающихся тканей, тем чувствительнее они к тера- тогенным агентам. Необходимо учитывать, что повреждающий агент может обладать боль- шим или меньшим сродством к тем или иным тканевым зачаткам, что обус- ловливает иногда некоторые специфические черты, характерные для опреде- ленного патогенного агента. Так, с 1957 по 1964 г. в ФРГ и других странах имела место так называемая талидомидная катастрофа. Талидомид применялся в качестве успокаивающего (снотворного) средства. Оказа- лось, что малые дозы этого препарата опасны для человеческого эмбриона; на животных они не действуют. У многих женщин, принимавших талидомид на II месяце беременности, рождались дети с тяжелыми пороками развития конечностей — амелией, фокомелией. В 40 % случаев пора- жались верхние конечности, в 10% —нижние, в 20% —верхние и нижние конечности, в 20% — конечности (верхние и нижние), органы слуха и зрения (данные 1961 и 1962 гг.). По данным 1964 г., в 45 % случаев талидомидные эмбриопатии протекали с пороками развития внутренних орга- нов. Из приведенного наблюдения видно, что талидомид имеет особый тропизм к развиваю- щимся закладкам конечностей. Кроме нарушений морфогенеза, удалось показать, что у эмбриона могут наблюдаться резорбция его некротизированных тканей, отек тканей, кровоиз- лияния и в конце эмбриогенеза даже неполная регенерация с образованием рубцов. Следует учитывать, что отмирание тканевых зачатков наблюдается и при нормальном ходе морфогенеза, например при слиянии отдельных за- чатков, образовании полостей в них, разрывах мембран (глоточной, клоакаль- ной) и др. Однако по объему и характеру процесс физиологического отмира- ния клеток отличается от некрозов в условиях патологии, он не сопрово- ждается рубцеванием, а главное, не приводит к нарушению процессов формирования. Обширные некрозы тканей эмбриона с рубцеванием по- являются, вероятно, при эмбриопатиях, обусловленных действием экзогенных агентов. При генотипических пороках развития значительной альтерации за- 517 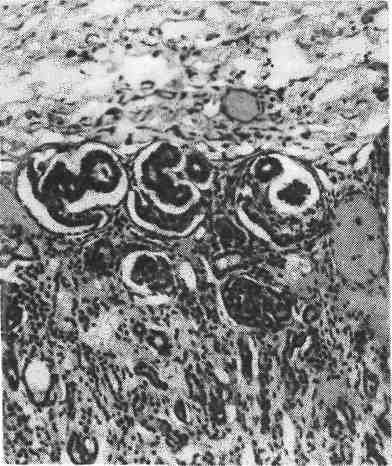 Рис. 372. Клубочки зародышевого типа в коре почки у мальчика в возрасте 7 дней. чатков органов, вероятно, не бывает, а имеется задержка процессов диф- ференцировки зачатков. Конкретных данных по патогене- зу наследственно обусловленных по- роков развития человека пока очень мало. Указанная разница в патогене- зе генетических и экзогенных поро- ков развития лишь предположитель- ная. В подавляющем большинстве случаев морфология сложившегося порока развития неспецифична. По- этому отличить по внешнему виду генотипический порок от фенокопии1 невозможно. Осно- вным проявлением патоло- гии эмбрионального пе- риода является дизонтоге- нез в виде врожденных пороков развития органов или частей тела заро- дыша. К фетальному периоду основной органогенез заканчивается и происходят дальнейший рост и дифференцировка тканей плода. В раннем фетальном периоде еще продолжается органогенез полушарий большого мозга, поэтому в этом периоде могут возникать пороки развития головного мозга. Кроме дизонтогенеза, у плода иногда встречаются и другие патологиче- ские процессы, так как его реактивные возможности по сравнению с эмбрио- ном возрастают. У плода наблюдаются альтеративные изменения, редуциро- ванное воспаление (см. «Воспаление»), иммуноморфологические изменения (см. «Иммунопатологические процессы», с. 143), расстройства крово- и лим- фообращения, гиперплазия и регенерация. Поэтому в фетальном периоде на- блюдаются болезни, сходные с болезнями внеутробного периода. Для болез- ней плода —фетопатий — характерны следующие особенности.
1 Фенокопия — порок развития, возникающий под влиянием экзогенных агентов, морфоло- гически идентичный генотипическому пороку. 518 Пороки, гены которых локализованы в Х-хромосоме, в свою очередь могут наследоваться по рецессивному или доминантному типу. Пороки, сцепленные с Х-хромосомой, передающиеся по рецессивному типу, наблюдаются как правило у мальчиков, так как единственная у них X- хромосома является пораженной. Мутантный ген передает мать, не являющаяся больной. Очень редко носительницей порока может быть девочка. Это бывает в том случае, если отец являлся больным, а мать — носительницей мутантного гена. Кроме локального поражения генетического аппарата ядра гаметы вслед- ствие мутации генов, в период гаметогенеза может появляться мутация хро- мосом в виде изменений их числа и структуры. Мутации хромосом получили название хромосомных аберраций. Хромосомные аберрации возни- кают чаще всего в момент редукционного деления гамет. Их следствием являются хромосомные болезни, которые, однако, в большинстве случаев не наследуются, так как их носители чаще умирают в детстве или являются бесплодными. Типичными примерами хромосомных болезней являются болезнь Дауна (трисомия по 21-й паре аутосом), синдром Патау (трисомия по 13—15-й паре аутосом), синдром Шерешевского- Тернера (моносомия половой хромосомы — 45 ХО) и др. Наиболее часто встречается болезнь Дауна, наблюдающаяся у новорожденных в соот- ношении 1 : 600, 1 : 700. Клинически у детей с рождения отмечается выраженная задержка ум- ственного и физического развития. Больные имеют типичный внешний вид — косой разрез глаз, западающая спинка носа, высокое небо, низкое расположение маленьких ушных раковин, выра- женная гипотония мышц. Дети редко доживают до возраста взрослого человека и погибают ча- ще от интеркуррентных заболеваний. У большинства из них обнаруживаются пороки развития сердца и магистральных сосудов (тетрада Фалло и др.), реже — пороки развития пищеваритель- ной и мочеполовой систем. У этих детей отмечаются недоразвитие полушарий большого мозга, особенно лобных его долей с задержкой дифференцировки нейронов, нарушения процессов мие- линизации, архитектоники кровеносных сосудов мозга. Синдром Патау у новорожденных и мертворожденных встречается с частотой 1 : 5149 рождений. Характерны выраженная общая гипоплазия, аномалии черепа и лица — низкий скошенный лоб, узкие глазные щели, запав- шее переносье, широкое основание носа, гипотелоризм, «дефекты скальпа», низко расположенные деформированные ушные раковины, типичны расще- лины верхней губы и неба. Отмечается полидактилия и флексорное положение кистей, микрофтальмия, колобома и помутнение роговицы. Со стороны го- ловного мозга отмечается микроцефалия, аринэнцефалия (отсутствие обоня- тельного мозга), аплазия или гипоплазия червя мозжечка и др. Отмечаются также врожденные пороки сердца, органов пищеварения, мочевой системы и др. Дети нежизнеспособны. Бластопатии Бластопатия — патология бластоцисты, возникающая в период нида- ции и дробления в первые 15 дней от момента оплодотворения до выделения эмбрио- и трофобласта. Этиология и патогенез. Причиной бластопатии чаще всего являются хромо- сомные аберрации в сочетании с влияниями среды (эндокринными заболева- ниями матери, гипоксией и др). Патогенез зависит от вида поражения бласто- цисты. Так, например, патогенез двойниковых уродств связан с появлением во время дробления двух или более самостоятельно растущих центров. Пола- гают, что если эти центры разобщены друг с другом, то развиваются два н е - зависимо растущих однояйцевых близнеца, нормальное развитие ко- торых не следует относить к бластопатиям. Если центры роста расположены близко и имеют общую для двух близнецов промежуточную зону, то разви- ваются два сросшихся близнеца. В обоих случаях возможно развитие симметричных и асимметричных близнецов (рис. 373). 520 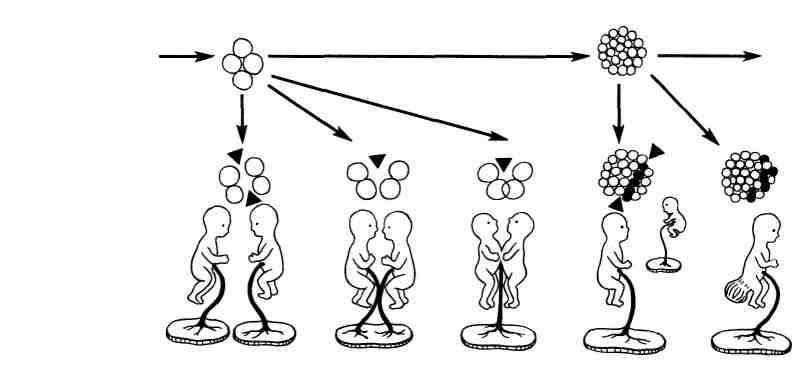 Рис. 373. Схематическое изображение возможных нарушений развития в период бласто- генеза (по Гертлеру). Морфология бластопатий разнообразна. К ним относятся нарушения им- плантации бластоцисты, а именно: эктопическая беременность, поверхностная или очень глубокая имплантация бластоцисты в эндометрий, нарушение ориентации формирующегося эмбриобласта в бластоцисте по отношению к эндометрию, аплазия или гибель развивающегося эмбриобласта с образова- нием пустого зародышевого мешка, пороки развития всего эмбриона, неко- торые одиночные пороки, двойниковые уродства и, наконец, аплазия или ги- поплазия формирующегося трофобласта — амниона, амниотической ножки, желточного мешка (В. П. Кулаженко). Поверхностная или чрезмерно глубокая имплантация бластоцисты приводит к порокам разви- тия формы, локализации, а также приращению плаценты (см. с. 541), которые чреваты гибелью плода во время акта родов. Нарушения ориентации эмбриобласта при полной топографической инверсии заканчиваются ги- белью эмбриобласта. При неполной инверсии наблюдаются пороки развития пуповины (см. с. 541), которые могут приводить кчгибели плода во время ро- дов. Пустые зародышевые мешки представляют собой бластоцисты, не содержащие эмбриобласт или содержащие его остатки. Иногда в них мож- но обнаружить амниотические оболочки, пуповину, желточный мешок. Патология развития всего эмбриона представляет собой об- щие тяжелые нарушения, несовместимые с жизнью. Одиночные и множественные пороки развития, возникаю- щие в период бластулы (в первые 8—12 нед), встречаются в 14,3 — 22,9% всех спонтанно абортированных зародышей. При этом в 46,2 % случаев они сопро- вождаются аномалиями последа. Такое сочетание часто приводит к гибели зародыша. Двойниковые уродства встречаются в виде сросшейся двойни. Если срос- шаяся двойня состоит из равных симметрично развитых компонентов, она на- зывается диплопагусом (diplopagus от греч. diplos — двойной, pagus — соединять); если же она состоит из асимметрично развитых компонентов — гетеропагусом (heteropagus от греч. heteros — другой), при этом недоразвитый близнец, находящийся в зависимости от другого, раз- витого, получил название паразита. К асимметричным двойниковым срос- шимся уродствам некоторые исследователи относят тератомы. Для обо- значения локализации сращения близнецов к анатомическому названию места 521 сращения добавляют также слово пагус; например, сращение в области го- ловы называют краниопагусом, в области груди — торакопагусом, в области таза —ишиопагусом и др. Двойниковые уродства сочетаются с нежизнеспособностью, в редких слу- чаях описана значительная продолжительность жизни таких близнецов до зре- лого возраста. В легких случаях сращений только мягких тканей возможна хирургическая коррекция. Эмбриопатии Эмбриопатия — патология эмбрионального периода с 16-го дня бере- менности до 75-го дня включительно, в течение которого заканчивается ос- новной органогенез и формирование амниона и хориона. К основным видам эмбриопатии относят врожденные пороки развития. Врожденным поро- ком развития называют стойкое морфологическое изменение органа, ча- сти тела или всего организма, выходящее за пределы вариаций нормального строения определенного биологического вида, возникающее внутриутробно в результате нарушений морфогенеза. Так как органогенез завершается в ос- новном в эмбриональный период, большинство пороков развития появляется именно на этом этапе внутриутробного существования. Однако, кроме вро- жденных пороков с нарушениями основного морфогенеза органов или частей тела, имеются врожденные пороки, при которых нарушения развития наблю- даются на уровне тканевой дифференцировки. Они часто бывают системны- ми, например пороки развития поперечнополосатой мускулатуры (врожден- ная миатония Оппенгейма), соединительной ткани (болезнь Марфана), кожи (врожденный ихтиоз), костей хрящевого генеза (врожденная хондродисплазия) и др. Пороки развития могут касаться также тканей одного органа, например гипоплазия гладкой мышечной ткани при megaureter, нервного интрамураль- ного аппарата — при megacolon, легочной ткани — при кистозном легком и др. По срокам возникновения эти пороки относятся к ранним фетопатиям. Ран- ние фетопатии часто сочетаются с эмбриопатиями; например, врожденный ихтиоз и хондродисплазия — с пороками развития лица, болезнь Марфана — с пороками развития лица и аорты и др. Частота врожденных пороков, по данным ВОЗ, составляет 1,3 % от общего числа рождений. Любой врожденный порок может проявляться в виде: 1) отсутствия како- го-либо органа или части тела (агенезия, аплазия); 2) недоразвития органа (гипоплазия); 3) чрезмерного развития (гиперплазия) или наличия избыточно- го числа органов (удвоение и др.); 4) изменения формы (слияние органов, атрезия, стеноз отверстий, каналов, дизрафия — незаращение эмбриональных щелей, экстрофия — выворот и др.; 5) изменения в расположении органов (эк- топия); 6) персистирование эмбриональных провизорных (предсуществовав- ших) органов. Классификация. Врожденные пороки развития разделяют по степени рас- пространенности в организме, по локализации в том или ином органе, по этиологии. По распространенности врожденные пороки могут быть: 1) изолированными — с поражением одного органа; 2) системными — с пора- жением нескольких органов одной из систем; 3) множественными — с пораже- нием органов разных систем. По локализации различают пороки развития центральной нервной, сердечно-сосудистой, пищеварительной, мочеполовой и других систем. Врожденные пороки развития названной локализации имеют наибольшее значение в патологии. Чаще всего встречаются пороки развития центральной нервной и сердечно-сосудистой систем, так как именно эти си- стемы имеют наибольший тератогенный терминационный период (см. рис. 371). Изолированные пороки развития встречаются чаще множественных, несмотря 522 на то что тератогенный терминационный период для многих органов во вре- мени совпадает. Наиболее совершенной является классификация пороков развития п о Этиологии, однако уровень современных знаний пока не позволяет ее при- держиваться. Известны лишь отдельные виды системных и множественных врожденных пороков, связанных с определенной этиологией, например рубео- лярная эмбриопатия, алкогольная, талидомидная эмбриопатия и др., а также наследственно обусловленные генотипические врожденные пороки и вро- жденные пороки вследствие хромосомных аберраций; последние, как правило, носят характер множественных. .>; Разграничение генотипических врожденных пороков от их фенокопий возможно с помощью генеалогического метода изучения родословной, цитогенетического метода, позволяющего изучить кариотип тканей носителя порока при их культивировании, с помощью близнецового метода, основанного на частоте выявления врожденных пороков у однояй- цевых близнецов и метода дерматоглифики — изучения комплекса кожных узоров, рас- положенных на ладонях, подошвах и сгибательной поверхности пальцев, который используется для срочной диагностики хромосомных болезней. |