учебник по патфизу. Учебник для слушателей и курсантов военномедицинской академии и военномедицинских институтов под редакцией проф. В. Ю. Шанина
 Скачать 4.96 Mb. Скачать 4.96 Mb.
|

|
Глава 15. ПАТОЛОГИЯ КЛЕТКИ Учение о повреждении клетки в современной медицине имеет особое значение. Это обусловлено, по меньшей мере, тремя обстоятельствами: во первых, оно тесно связано с разработкой вопросов возникновения, развития и исходов болезней, поскольку любой патологический процесс сопровождается повреждением клетки; во вторых, все более интенсивное внедрение в клиническую практику различных способов восстановления жизнедеятельности поврежденных органов и тканей поставило задачу по исследованию механизмов устранения или уменьшения степени их альтерации, а также разработку методов активации защитных, компенсаторных и приспособительных реакций в клетках с целью оптимизации процесса выздоровления; в третьих, понимание многих открытий молекулярной патологии последнего времени становится возможным лишь при условии определения их места и значения с позиций патологии клетки и межклеточного взаимодействия. Все эти моменты касаются и военной медицины. Так как любое боевое ранение и повреждение сопровождается гибелью и патологическими изменениями в клетках. Клетка является элементарной саморегулирующейся структурно-функциональной единицей тканей и органов. В ней протекают процессы, лежащие в основе энергетического и пластического обеспечения меняющихся структур и уровня функционирования тканей и органов. Различные патогенные агенты, действующие на клетку, могут обусловить ее повреждение. Под повреждением клетки понимают такие изменения ее структуры, метаболизма, физико-химических свойств и функций, которые ведут к нарушению жизнедеятельности. В учении о повреждении клетки условно выделяют три раздела:
Роль клеточных повреждений в развитии патологии и их причины При возникновении повреждений в организме формируются общие и местные реакции. К числу общих реакций организма относятся: стресс, шок, кома, боль, лихорадка, активация протеолитических систем. Местные повреждения связаны с повреждением клетки и ее структуры. Первая научная теория о роли клеточных нарушений в патологии организма была сформулирована немецким патологом Рудольфом Вирховым, который главным в любой болезни человека считал повреждение клеток соответствующих органов. Действительно, клетка является элементарной частицей живых систем любого уровня, но и сама представляет собой саморегулирующую систему. Вот почему повреждение клетки или группы клеток, нарушая естественно их интеграцию и кооперацию в органе и организме, является материальной основой развивающейся патологии. Однако, хотя роль клеточных повреждений является по сути, базой любого заболевания, все мы рассматриваем болезнь, как страдание целостного организма, поскольку с закономерной неизбежностью в патологию вовлекается весь организм, как единое целое. Причины повреждения клетки Повреждение клетки может быть результатом воздействия на нее множества агентов. Эти агенты подразделяют на различные группы в зависимости от их природы и происхождения. Виды причин повреждения клетки по их природе. Физические факторы. К наиболее частым причинам повреждения клетки физической природы относятся следующие:
Патогенное действие на клетку могут также оказывать электромагнитные и другие физические факторы. Химические факторы. К числу факторов повреждения клетки химической природы относятся разнообразные вещества: органические и неорганические кислоты, щелочи, соли тяжелых металлов, продукты нарушенного метаболизма. Так, цианиды подавляют активность цитохромоксидазы. Этанол и его метаболиты ингибируют многие ферменты клетки. Вещества, содержащие соли мышьяка, угнетают пируватоксидазу. Неправильное применение лекарственных средств также может привести к повреждению клеток. Например, передозировка строфантина обуславливает значительное подавление активности К+, Na+ - АТФазы сарколеммы клеток миокарда, что ведет к дисбалансу интрацеллюлярного содержания ионов и жидкости. Передозировка инсулина может вызвать быстрое использование глюкозы, истощение ее запаса в виде гликогена и нарушение энергетического обеспечения клетки. Важно, что повреждение клетки может быть обусловлено как избытком, так и недостатком одного и того же агента. Например, избыточное содержание кислорода в тканях активирует процесс свободнорадикаьного перекисного окисления липидов (СПОЛ), продукты которого повреждают ферменты и мембраны клеток. С другой стороны, снижение содержания кислорода обусловливает нарушение окислительных процессов, понижение образование АТФ и, как следствие, расстройство функций клетки. Биологические факторы. К этой группе причин повреждения клетки относится большое число факторов. Наибольшее значение среди них - вирусы, риккетсии, бактерии, паразиты, грибы. Продукты их жизнедеятельности или деградации вызывают расстройства функций клетки, нарушают течение в ней метаболических реакций, проницаемость или даже целостность мембран, подавляют активность клеточных ферментов. Повреждение клетки нередко обусловливается факторами иммунных и аллергических процессов. Они могут быть вызваны, в частности, сходством антигенов, например, микроорганизмов и клеток организма. Так, некоторые разновидности гемолитического стрептокока и белки базальной мембраны клубочков почек имеют близкий антигенны состав. Попадая в организм, они вызывают образование антител, повреждающих как стрептококки, так и нефроны. Эндо- и экзотоксины, а также структурные компоненты бактерий, вирусов и паразитов могут изменить антигенный состав клеток, что приводит к выработке антител или иммунных Т-лимфоцитов, повреждающих собственные клетки, в результате чего может развиться аутоиммунный патологический процесс. Повреждение может быть также результатом образования антител или Т-лимфоцитов, действующих против неизмененных клеток организма вследствие мутаций в геноме В- или Т-лимфоцитов иммунной системы. Важную роль в поддержании метаболических процессов в клетке играют физиологически активные вещества, а также факторы, поступающие из окончаний нейронов, в частности ферменты, белки, липиды, адениннуклеотиды, микроэлементы и др. Их дефицит или избыток может стать причиной расстройств обмена веществ в клетках, нарушения их жизнедеятельности и развития различных патологических состояний. Повреждение клеток нередко бывает обусловлено значительно повышенной функцией органов и тканей. Например, при длительной чрезмерной физической нагрузке возможно развитие сердечной недостаточности в результате нарушения жизнедеятельности кардиомиоцитов. Виды причин повреждения клетки по их происхождению. По происхождению причинные факторы повреждения клетки делят на 2 подгруппы:
Экзогенные факторы. К ним относятся физические воздействия: механические, электрический ток, тепло, холод; химические агенты; биологические: вирусы, риккетсии, бактерии, паразиты и др. Эндогенные факторы. К ним относятся агенты физической природы: избыток в клетке или во внеклеточной среде свободных радикалов, значительные колебания осмотического давления; химические факторы: избыток и дефицит ионов (К+, Н+,Са2+ и др.), кислорода, углекислого газа, перекисных соединений органического и неорганического характера, метаболитов и др; факторы биологической природы: дефицит или избыток физиологически активных веществ (катехоламинов, гормонов, простагландинов и др.); продукты жизнедеятельности или распада вирусов, бактерий, паразитов, риккетсий; продукты, высвобождающиеся из других поврежденных или погибших клеток. Инфекционные факторы. Примерами их могут быть токсины микроорганизмов, паразитов и они сами. Факторы неинфекционного генеза. К ним относятся факторы физической, химической или биологической природы немикробного генеза. Действие повреждающих факторов на клетки осуществляется прямо или опосредовано. В последнем случае речь идет о формировании цепи вторичных реакций, образование веществ-посредников, реализующих повреждающее действие так называемого "первичного" патогенного фактора. Его действие может опосредоваться через изменения нервных или эндокринных воздействий на клетки (например, при стрессе, шоке); нарушение системного кровообращения (при сердечной недостаточности); отклонение физико-химических параметров (при состояниях, сопровождающихся ацидозом, алкалозом, образованием свободных радикалов, продуктов СПОЛ, дисбалансом ионов и жидкости); иммуноаллергические реакции при аутоаллергических заболеваниях; образование избытка или недостаток БАВ (гистамина, кининов, простагландинов). Многие из этих и других, участвующих в развитии различных форм патологии клетки, получили название посредников, или медиаторов (например, медиаторы воспаления, аллергии, канцерогенеза и др.), повреждения. Общие механизмы повреждения клеток Механизмы клеточных повреждений при действии различных этиологических факторов тесно взамосвязаны. Сразу подчеркнем, что любой этиологический фактор обусловливает, как правило, несколько механизмов повреждения. Вот почему изолированное выделение отдельных механизмов повреждения клетки оправдано лишь с точки зрения удобства их рассмотрения и понимания. На уровне клетки повреждающие факторы "включают" несколько патогенетических звеньев. К числу основных относят: Нарушение энергетического обеспечения процессов, происходящих в клетке Этот механизм часто является инициальным и ведущим в альтерации клетки. Энергоснабжение может рассматриваться на этапах ресинтеза АТФ, транспорта, а также утилизации энергии АТФ. Ресинтез АТФ нарушается в результате дефицита кислорода и (или) субстратов метаболизма, снижения активности ферментов тканевого дыхания и гликолиза, повреждения и разрушения митохондрий, в которых осуществляются реакции цикла Кребса и перенос электронов к молекулярному кислороду, сопряженный с фосфорилированием АДФ. Заключенная в макроэргических связях АТФ энергия в норме доставляется от мест ее синтеза (из митохондрий и гиалоплазмы) к эффекторным структурам (миофибриллам, мембранным ионным "насосам" и др.) с участием ферментных систем АДФ-АТФ-транслоказы (адениннуклеотидтрансферазы) и креатининфосфокиназы (КФК). Адениннуклеотидтрансфераза обеспечивает транспорт энергии макроэргической фосфатной связи АТФ из матрикса митохондрий через их внутреннюю мембрану, а КФК - далее на креатин с образованием креатинфосфата, который поступает в цитозоль. КФК эффекторных клеточных структур транспортирует фосфатную группу креатинфосфата на АДФ с образованием АТФ, который и исполь 3зуется в процессах жизнедеятельности клетки. Ферментные системы транспорта энергии также могут быть повреждены различными патогенными агентами, в связи с чем даже на фоне высокого общего содержания АТФ в клетке может развиваться его дефицит (гипоэргоз клетки) в энергорасходующих структурах. Нарушение энергообеспечения клеток и расстройство их жизнедеятельности может развиваться в условиях достаточной продукции и нормального транспорта энергии АТФ. Это может быть 3 результатом повреждения механизмов утилизации энергии главным образом за счет снижения активности АТФаз (АТФазы актомиозина, К+, Na+-зависимой АТФазы плазмолеммы, Mg2+-зависимой АТФазы "кальциевой помпы" саркоплазматической сети и др.). Следовательно, расстройство жизнедеятельности клеток может развиться даже в условиях нормального или повышенного содержания в клетке АТФ. Нарушение энергообеспечения в свою очередь может стать одним из факторов расстройств функции мембранного аппарата клеток, их ферментных систем, баланса ионов и жидкости, а также механизмов регуляции клетки. Так патогенетическая роль ионов кальция в организме впервые показана в 1883 году английским биохимиком Рингером, который установил, что изолированное сердце лягушки, перфузируемое раствором, не содержащим ионов кальция, прекращает свою работу. Это был первый факт, демонстрирующий значение ионов кальция в сократительной функции мышечной клетки сердца. Вскоре выявили, что при недостатке Са2+ во внеклеточной жидкости происходит:
Позднее стали известны механизмы нарушения клеточных функций, связанные с избытком Са2+ во внутриклеточном пространстве. Механизмы повышения концентрации Са2+ внутри клетки. В норме внутри клетки концентрация Са2+ (ионизированного) составляет 2х10-7 моль/л, а вне ее - приблизительно в 5000 раз больше - 1х10-3 моль/л. Причем концентрация Са2+ в митохондриальном пуле в 500 раз выше, чем в цитоплазме, что свидетельствует о существовании механизмов внутриклеточной регуляции его содержания. Высокая разность концентрации Са2+ по обе стороны цитоплазматической мембраны предполагает различие механизмов его поступления и выведения из клетки: поступление его в клетку - это в значительной степени пассивный процесс, но его выведение осуществляется активно с помощью Са2+-зависимой АТФ-азы. Поэтому избыточная концентрация Са2+ в клетке может быть обусловлена двумя факторами: увеличением поступления и/или нарушения работы Са2+-насосов, выталкивающих его наружу. Механизмы увеличенного поступления Са2+ внутрь клеток Увеличенный приток Са2+ в клетку может происходить как через поврежденную, так и через неповрежденную мембрану. Причем в первом случае его концентрация в клетке значительно нарастает, что является характерным признаком погибающей клетки. При относительно небольших мембранных повреждениях возможно формирование особых каналов, ионофоров, через которые ионы Са2+ могут поступать внутрь клетки, способствуя ее гибели. Если целостность мембран не нарушена, то Са2+ попадает в клетку через вида каналов:
Причины нарушения удаления кальция из клетки В основе этих нарушений лежит повреждение энергозависимых мембранных насосов:
Все это дает картину неспецифического набухания митохондрий. Механизмы повреждения клетки кальцием Избыток Са2+ в клетке вызывает следующие нарушения ее структуры и функции:
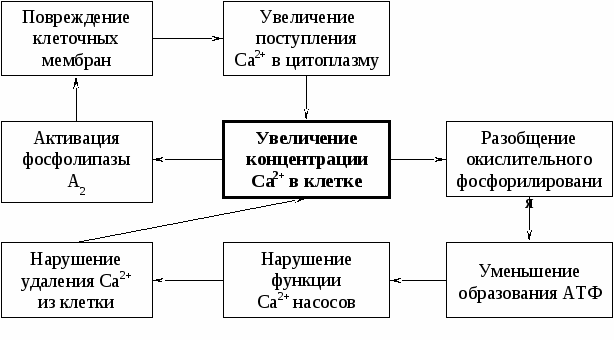 Рис. 15.1. Порочные круги при повреждении клетки ионами кальция Механизмы предупреждения кальциевых повреждений клеток Они направлены на:
Патогенетические принципы терапии кальциевых повреждений клетки
Повреждения мембранного аппарата и ферментных систем клетки Этот механизм играет существенную роль в расстройстве жизнедеятельности клетки, а также в переходе обратимых изменений в ней в необратимые. Это обусловлено тем, что основные свойства клетки в существенной мере зависят от состояния ее мембран и энзимов. Согласно модели мембраны клеток, предложенной S. Singer и G.Nicolson (1972), она представляет собой вязкую полужидкую мозаичную структуру. Основу ее составляют молекулы фосфолипидов (липидная фаза мембраны), полярные (ионные) "головки" которых направлены к водной среде, т.е. к гидрофильным поверхностям мембран, а неполярные части ("хвосты") - внутрь них (гидрофобная зона). В фосфолипидной среде взвешены белковые молекулы, часть из которых полностью погружена в мембраны и пронизывает их толщу (так называемые интегральные белки), а часть расположена на их поверхности ("периферические" белки). Периферические белки не проникают в толщу мембраны и удерживаются на ее поверхности главным образом электростатическими силами. Белковые молекулы могут менять занимаемое ими положение в липидной фазе мембраны, что влияет на интенсивность и характер протекания катализируемых ими реакций. Кроме того, липиды мембран нередко обеспечивают оптимальные условия для энзиматических процессов. Например, окислительное фосфорилирование требует безводной среды, что предотвращает "спонтанный" гидролиз АТФ. В последние годы представления о структуре мембран дополнены положением о том, что ее компоненты (белки, гликопротеиды, гликозоаминогликаны, гликолипиды) взаимодействуют между собой, а также с микрофиламентами, микротрубочками, тонофибриллами цитоплазмы клеток, образуя целостную динамическую систему - твердоэластический каркас. Этот каркас "вмонтирован" в жидкую липидную фазу мембран. Наличие каркаса обеспечивает относительно стабильное расположение на (в) мембране "антигенов", рецепторов, ферментов, и других ее компонентов, а также препятствует агрегации белков мембраны, которая была бы неизбежной при свободном движении их молекул в жидкой липидной среде. К числу основных механизмов повреждения мембран клеток относят:
Важно, что все указанные механизмы прямо или опсредованно обуславливают повреждение, изменение конформации и (или) кинетических свойств ферментов клетки, многие из которых связаны с мембранами. Одним из важнейших механизмов повреждения мембран и ферментов является чрезмерная активация свободнорадикальных реакций и СПОЛ. Эти реакции протекают в клетках и в норме, являясь необходимым звеном таких жизненно важных процессов, как транспорт электронов в цепи дыхательных ферментов, синтез простагландинов и лейкотриенов, пролиферация и созревание клеток, фагоцитоз, метаболизм катехоламинов и др. Реакции СПОЛ участвуют в процессах регуляции липидного состава биомембран и активности ферментов. Последнее является результатом как прямого действия продуктов липопероксидных реакций на энзимы, так и опосредованного - 3 через изменения состояния мембран, с которыми ассоциированы многие ферменты. Интенсивность СПОЛ регулируется соотношением факторов, активирующих (прооксиданты) и подавляющих (антиоксиданты) этот процесс (схема 2). К числу наиболее активных прооксидантов относятся легко окисляемые соединения, индуцирующие сводные радикалы, в частности 2 нафтохиноны, витамины А и D, восстановители НАДФН2, НАДН2, липоевая кислота, продукты метаболизма простагландинов и катехоламинов. В реакции пероксидации могут вовлекаться соединения различного биохимического состава: липиды, белки, нуклеиновые кислоты. Однако ведущее значение среди них имеют фосфолипиды. Это определяется тем, что они являются основным компонентом мембран и легко вступают в оксигенные реакции. Процесс СПОЛ условно можно разделить на три этапа:
Инициальным звеном свободнорадикальных перекисных реакций при повреждении клетки является, как правило, образование в процессе оксигенных реакций так называемых активных форм кислорода: синглетного (1О2), супероксидного радикала кислород (О27), перекиси водорода (Н2О2), гидроксильного радикала (.ОН). О27 генерируется лейкоцитами (особенно интенсивно при фагацитозе), в митохонддриях в процессе окислительных реакций, в тканях при метаболической трансформации катехоламинов, синтезе простагландинов и других соединений. Н2О2 образуется при взаимодействии (дисмутации) радикалов О27 в гиалоплазме клеток и матриксе митохондрий. Этот процесс может катализироваться ферментом супероксиддисмутазой (СОД): СОД О27 + О27 + 2Н+ = Н2О2 + О2 О27 и Н2О2 обладают повреждающим действием и сами по себе. Однако под влиянием ионов железа, присутствующих как в гиалоплазме клеток, так и в биологических жидкостях (межклеточной, плазме крови, лимфе), О27 и Н2О2 могут "трансформироваться" в весьма "агрессивный", обладающий высоким патогенным действием гидроксильный радикал .ОН: каталаза Н2О2 + Fe2+ = Fe3+ + ОН- + .ОН; О27 + Н2О2 = О2 + ОН- + .ОН; Радикалы .ОН активно вступают в реакции с органическими соединениями, главным образом липидами, а также нуклеиновыми кислотами и белками. В результате образуются их активные радикалы и перекиси. При этом реакция может приобрести цепной "лавинообразный" характер. Однако, это происходит не всегда. В клетках протекают процессы и действуют факторы, которые ограничивают или даже прекращают свободнорадикальные и перекисные реакции, т.е. оказывают антиоксидантный эффект. Одним из таких процессов является, в частности, взаимодействие радикалов и гидроперекисей липидов между собой, что ведет к образованию "нерадикальных" соединений. Ведущую роль в системе антиоксидантной защиты клеток играют механизмы ферментной, а также неферментной природы, главные из которых представлены в таблице 15.1. Таблица 15.1 Звенья антиоксидантной системы и ее некоторые факторы
Исследования последних лет показали, что чрезмерная интенсификация свободнорадикальных и перекисных реакций является одним из главных факторов повреждения мембран и ферментов клеток. Ведущее значение при этом имеют следующие процессы:
В норме состав и состояние мембран модифицируются не только свободнорадикальными липопероксидными процессами, но также мембраносвязанными, свободными (солюбилизированными) и лизосомальными ферментами: липазами, фосфолипазами, протеазами. Под влиянием патогенных факторов их активность или содержание в гиалоплазме клетки могут значительно повыситься (в частности, вследствие развития ацидоза, способствующего увеличению выхода ферментов из лизосом и их последующей активации). В связи с этим интенсивному гидролизу подвергаются глицерофосфолипиды и белки мембран, а также ферменты клеток. Это сопровождается значительным повышением проницаемости мембран и снижением кинетических свойств ферментов. В результате активации липопероксидазных реакций и гидролаз (главным образом липаз и фосфолипаз) в клетке накапливаются гидроперекиси липидов, свободные жирные кислоты, лизофосфолипиды, в частности глицерофосфолипиды, фосфатидилхолины, фосфатидилэтаноламины, фосфатидилсерины. Эти соединения получили название амфифильных в связи с их способностью проникать и фиксироваться в обеих (как гидрофобной, так и гидрофильной) средах мембран клеток (от греч. ampho - "оба", два, двояко). При сравнительно небольшом уровне в клетке амфифилиных соединений они, внедряясь в биомембраны, изменяют нормальную последовательность глицерофосфолипидов, нарушают структуру липопротеидных комплексов, увеличивают проницаемость, а также меняют конфигурацию мембран в связи с "клинообразной" формой липидных мицелл. Накопление в большом количестве амфифилов сопровождается массированным внедрением их в мембраны, что, также как и их избыток гидроперекисей липидов, ведет к формированию кластеров и микроразрывов в них. Потенцированию повреждения клеточных мембран в связи с торможением процессов обновления их компонентов и устранением дефектов в них способствует также расстройство энергетического "обеспечения" пластических процессов в клетке. Это обусловлено нарушением реакций репаративного ресинтеза поврежденных или утраченных липидных, белковых, липопротеидных, гликопротеидных и других молекул мембран, а также синтеза их заново значительные изменения физико-химического состояния мембран клеток могут быть обусловлены и модификацией конформации (пространственной структуры, формы) макромолекул белка, липопротеидов, гликопротеидов и других соединений. Причиной этому могут стать дефосфорилирование ("деэнергизация) указанных молекул в основном в связи с нарушением прпоцессов энергообеспечения клеток. При этом наблюдается изменение вторичной и третичной структуры белков, конформации липопротеидов, а также подавление каталитической активности ферментов. Повреждение мембран вызывает набухание клеток (в том числе и их органелл) в связи с их гипергидрацией. Значительное увеличение объема клеток и субклеточных структур (митохондрий, эндоплазматической сети, ядра и др.) обусловливает перерастяжение и нередко разрывы их мембран. Последнее является следствием увеличения осмотического и онкотического давления в клетках. Это в свою очередь обуловлено избытком в них гидрофильных молекул органических соединений (молочной, пировиноградной кислоты, альбуминов, глюкозы и др.), а также ионов. Таким образом, видно, что повреждение мембран и ферментов клеток является одной из частых и главных причин нарушения жизнедеятельности клеток. Дисбаланс ионов и жидкости, изменение электрофизиологических свойств клетки. Как правило, нарушение трансмембранного распределения, а также внутриклеточного содержания и соотношения различных ионов развивается вслед за или одновременно с расстройствами энергетического обеспечения и сочетается с признаками повреждения мембран и ферментов клеток. В результате этого существенно изменяется проницаемость мембран для многих ионов. В наибольшей мере это относится к калию, натрию, кальцию, магнию, хлору, т.е. ионам, которые принимают участие в таких жизненно важных процессах, как возбуждение, его проведение, электромеханическое сопряжение и др. Как правило дисбаланс ионов накоплением в клетке натрия и потерей калия вследствие нарушения работы К-, Na-зависимой АТФазы плазмолеммы, увеличением содержания кальция (в частности в результате расстройства функционирования натрий-кальциевого ионообменного механизма клеточной мембраны, который обеспечивает обмен двух ионов натрия, входящих в клетку, на один ион кальция, выходящий из нее). Увеличение внутриклеточного содержания Na+, конкурирующего с Са2+ за общий переносчик, также препятствует выходу кальция из клетки. Нарушение трансмембранного распределения катионов сопровождается изменением содержания в клетке и анионов хлора, НСО3-, ОН- и др. Следствием дисбаланса ионов являются изменение мембранного потенциала покоя и действия, а также нарушение проведения импульса возбуждения. Эти изменения имеют важное значение, поскольку нередко являются одним из важных признаков наличия и характера повреждения клеток. Примером могут служить изменения электрокардиограммы при повреждении клеток миокарда, электроэцефалограммы - при нарушении структуры и функций нейронов головного мозга, электромиограммы - при изменениях в мышечных клетках. Нарушения внутриклеточного содержания ионов обусловливает изменение объема клеток вследствие дисбаланса жидкости. Он проявляется либо гипергидратацией, либо гипогидратацией клетки. Так, например, повышение содержания ионов натрия и кальция в поврежденных клетках сопровождается увеличением в них осмотического давления. В результате этого в клетках накапливается вода. Клетки при этом набухают, объем их увеличивается, что сопровождается растяжением и нередко микроразрывами цитолеммы и мембран органелл. Напротив, дегидратация клеток (например, при некоторых инфекционных заболеваниях, сопровождающихся потерей воды) характеризуется выходом из них жидкости и растворенных в ней белков (в том числе ферментов), а также других органических и неорганических водорастворимых соединений. Внутриклеточная дегидратация нередко сочетается со сморщиванием ядра, распадом митохондрий и других органелл. Нарушение генетической программы клетки и (или) механизмы ее реализации Основными процессами, ведущими к изменению генетической информации клетки, являются:
Помимо изменений в генетической программе, важным механизмом расстройства жизнедеятельности клеток является нарушение реализации этой программы главным образом в процессе клеточного деления при митозе или мейозе. Данных о патологии мейоза (при развитии половых клеток) на сегодняшний день еще недостаточно. Это обусловлено, в частности, трудностью получения биопсийного материала из семенника или яичников. Более разработаны вопросы патологии митоза. Выделяют три группы нарушения митоза:
Действие на генетический аппарат клетки повреждающих факторов различного характера весьма высокой интенсивности может обусловить гибель ее. К числу наиболее значимых процессов, вызывающих гибель клеток, можно отнести следующие:
Существует мнение, что в клетках имеется специальная программа, реализация которой приводит к необратимой деструкции генетического материала и клеточной гибели. Считают, что программа гибели клетки связана с наличием в ее геноме специальных генов - "убийц". Эти гены сформировались на ранних этапах эволюции многоклеточных организмов для элиминации необратимо поврежденных и (или) патологически функционирующих клеток, представляющих реальную или потенциальную опасность для всего организма. При этом элиминированные клетки замещались нормальными в связи с делением соседних неповрежденных клеток. Это и обеспечивало стабильность структуры и жизнедеятельности тканей, органов и в целом многоклеточного организма как системы. Наличие генетической программы гибели клеток объясняет многие феномены:
Пути формирования патологии при нарушении в системе вторичных клеточных посредников Расстройство внутриклеточных механизмов регуляции функции клеток. Это может быть результатом нарушений, развивающихся на одном или нескольких уровнях регуляторных механизмов:
Характеристика типовых форм повреждения клеток Повреждение клеток характеризуется развитием разнообразных изменений в них. Однако их можно объеденить в несколько групп:
Дистрофии (от лат. dys - нарушение, расстройство + греч. trophe - питаю) - это нарушения обмена веществ в клетках, сопрвождающиеся расстройствами их функций, пластических процессов и структурными изменениями, ведущими к нарушению их жизнедеятельности. Основными механизмами дистрофий являются:
К числу основных разновидностей клеточных дистрофий в зависимости от преимущественно нарушенного вида обмена веществ относят:
Диспротеинозы. Характеризуются изменением фихико-химических свойств белков клеток и как следствие нарушением их ферментативной и структурной функций. Наиболее часто диспротеинозы проявляются в виде зернистой, гиалиново-капельной и гидропической дистрофии. Нередко они представляют собой последовательные этапы нарушения обмена цитоплазматических белков, приводящих к некрозу клеток. При зернистой дистрофии в цитоплазме появляются гранулы (зерна) белка. Они образуются в результате инфильтрации (проникновения) его из межклеточной жидкости, трансформации углеводов и жиров в белки, распада (декомпозиции) липопротеидов цитоплазмы и мембран. Одной из главных общих причин зернистой дистрофии является нарушение энергообеспечения клеток. Гиалиновая дистрофия характеризуется накоплением в цитоплазме белковых гиалиноподобных ацидофильных включений ("капель"). Одновременно с этим выявляются признаки деструкции клеточных органелл. Признаки гиалиновой дистрофии наблюдаются при состояниях, вызывающих повышение проницаемости клеточных мембран. Гидропическая (водяночная, вакуольная) дистрофия является результатом такого изменения физико-химических свойств белков цитоплазмы, которое сопровождается повышением онкотического давления в клетке и избыточной гидратацией белковых мицелл. В цитоплазме клеток формируются вакуоли, наполненные жидкостью и не содержащие липидов или гликогена. При электронной микроскопии обнаруживаются признаки внутриклеточного отека и набухания органелл. Наиболее частыми причинами гидропической дистрофии являются гипоксия, воздействие ионизирующей радиации, токсины микроорганизмов и паразитов, нарушения питания. Липидозы. К липидозам относят различные по химическому составу вещества, нерастворимые в воде. Липидозы проявляются либо увеличением содержания внутриклеточных липидов, либо появлением их в клетках, где они в норме отсутствуют, либо образованием липидов аномального химического состава. Липидозы, так же как и диспротеинозы, наиболее часто наболюдаются в клетках сердца, печени, почек, мозга и носят соответсвующие названия (жировая дистрофия сердца, печени, почек, мозга). Углеводные дистрофии. Характеризуются нарушением обмена полисахаридов (гликогена, мукополисахаридов) и гликопротеидов (муцина, мукоидов). "Полисахаридные" дистрофии проявляются:
Причиной углеводных дистрофий чаще всего являются эндокринопатии (например, инсулиновая недостаточность) или ферментопатии (отсутствие или низкая активность ферментов, принимающих участие в процессах синтеза и распада углеводов). Углеводные дистрофии, связанные с нарушением метаболизма гликопротеидов, характеризуются, как правило, накоплением муцинов и мукоидов, имеющих слизистую консистенцию. В связи с этим их называют слизистыми дистрофиями. Причинами их наиболее часто служат эндокринные расстройства (например, недостаточная продукция или низкая активность гормонов щитовидной железы), а также прямое повреждающее действие на клетки патогенных факторов. Пигментные дистрофии (диспигментозы). Пигменты клеток организма человека и животных принимают участие в реализации многих функций: синтез и катаболизм веществ, рецепция различных воздействий, защита от повреждающих факторов. Клеточные пигменты являются хромопротеидами, т.е. соединениями состоящими из белка и красящего вещества. В зависимости от биохимического строения эндогенные клеточные пигменты разделяют следующим образом:
Все диспигментозы делятся на несколько групп в зависимости от их происхождения, механизма развития, биохимической структуры пигмента, проявлений и распространенности. Виды диспигментозов По происхождению:
По механизму развития:
По биохимической структуре пигмента:
По проявлениям:
По распространенности:
Гемоглобиногенные диспигментозы включают гемосидероз, гемохроматоз, гемомеланоз, порфирию, накопление избытка прямого билирубина в гепатоцитах. Большинство гемоглобиногенных пигментов относятся к продуктам катаболизма гемоглобина. Некоторые из них (ферритин, гемосидерин) образуются с участием железа, всасывающегося в кишечнике. |