Глик Молекулярная биотехнология. Глик Б., Пастернак Дж. Молекулярная биотехнология. Принципы и применение. Пер с англ. М. Мир, 2002. 589 с
 Скачать 9.74 Mb. Скачать 9.74 Mb.
|

Стабилизация белковОбычно время полужизни белков составляет от нескольких минут до нескольких часов. Такая вариабельность обусловливается различиями в числе дисульфидных связей в белковых молекулах и наличием или отсутствием на 5'-конце определенных аминокислот. Например, если к N-концу ß-галактозидазы присоединять разные аминокислоты, то время жизни модифицированного белка in vitro может варьировать от двух минут до более 20 часов (табл. 6.4). Аминокислоты, увеличивающие время жизни белков, можно включать в белки генноинженерными методами. Часто для стабилизации белка-мишени достаточно присоединить к N-концу всего
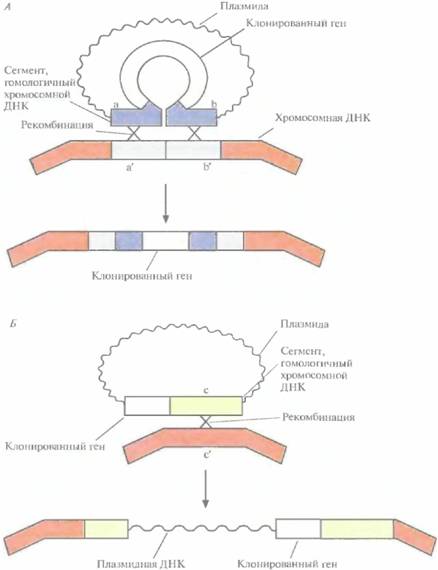
122 ГЛАВА 6 один аминокислотный остаток. Долгоживущие белки накапливаются в клетках, что увеличивает конечный выход продукта. Это характерно как для эу-, так и для прокариот. Однако стабильность белков может не только повышаться. Так, включение некоторых аминокислотных последовательностей во внутреннюю часть белковой молекулы делает ее более чувствительной к протеолитическому расщеплению. Такие последовательности обогащены остатками пролина (Р), глутаминовой кислоты (Е), се-рина (S) и треонина (Т), отсюда и их название -PEST-последовательности. Они часто бывают фланкированы кластерами из положительно заряженных аминокислот и, возможно, служат маркерами для протеаз. Стабильность белков, содержащих такие последовательности, можно было бы повысить, внося изменения в соответствующие гены. При этом, однако, необходимо позаботиться о том, чтобы не произошло нарушений функции белка-мишени. Рост в условиях недостатка кислородаE. coliи многие другие микроорганизмы, которые используются для экспрессии чужеродных белков, обычно растут только в присутствии кислорода. К сожалению, растворимость кислорода в водных средах ограничена, а по мере увеличения плотности культуры содержание растворенного кислорода в культуральной среде быстро падает. Более того, поскольку кислород растворяется очень медленно, эту проблему нельзя решить простым продуванием через среду воздуха или кислорода даже при интенсивном перемешивании. При уменьшении концентрации кислорода экспоненциальный рост замедляется и культура медленно переходит в стационарную фазу, характеризующуюся другим метаболическим статусом. Одним из последствий этого является образование в клетках протеиназ, которые могут расщеплять белок-мишень. Проблему аэрации культуральной среды пытались решить разными способами: изменением конструкции биореактора, повышением интенсивности продувания воздуха и перемешивания, добавлением в среду веществ, увеличивающих растворимость кислорода. Все это, однако, не привело ни к каким ощутимым результатам. Применение хозяйских штаммов с дефицитом протеиназОдин из возможных подходов к стабилизации чужеродных белков, синтезируемых E, coli, состоит в использовании хозяйских штаммов с дефицитом протеолитических ферментов. Однако здесь есть свои трудности. В клетках E. coli синтезируется по крайней мере 25 разных протеиназ, и только некоторые из них изучены на генетическом уровне. Кроме того, протеиназы выполняют в клетке очень важную функцию, разрушая чужеродные или дефектные белки и обеспечивая тем самым жизнеспособность клеток. В одной из работ были сконструированы штаммы, несущие мутации в одном или даже нескольких протеиназных генах, и чем более выражен был суммарный дефицит по протеиназам, тем хуже рос штамм. Таким образом, снижение протеиназной активности приводит к истощению клеточных ресурсов. И все же удалось создать штаммы E. coli, несущие мутации в гене сигма-фактора РНК-полимеразы, ответственного за синтез белков теплового шока (rpoН), и в гене протеиназы, необходимой для роста клеток при высоких температурах (degP), y которых удельная активность секретируемых белков была в 36 раз выше, чем у штаммов дикого типа. Это кажущееся увеличение было обусловлено снижением интенсивности протеолитического расшепления белков. Бактериальный «гемоглобин»Местообитанием некоторых штаммов грамотрицательных облигатных аэробных бактерий Vitreoscilla являются сильно обедненные кислородом непроточные водоемы. Чтобы получать нужное количество кислорода для роста и метаболизма, они синтезируют гемоглобиноподоб-ное вещество, связывающее кислород окружающей среды и увеличивающее концентрацию доступного кислорода в клетке. Когда ген, кодирующий этот белок, был введен в клетки E. coli, в последних сразу произошли серьезные изменения: повысился уровень синтеза клеточных и рекомбинантных белков, возросла эффективность протонных насосов, увеличилось количество образующегося АТР и его концентрация, особенно при низком содержании кислорода в среде. Чтобы такую стратегию можно было ис- Оптимизация экспрессии генов, клонированных в прокариотических системах 123 пользовать применительно к другим хозяйским клеткам, необходимо, чтобы эти клетки не только эффективно экспрессировали «гемоглобиновый" ген Vitreoscilla, но и синтезировали гем -составляющую гемоглобиновой молекулы. Это позволит улучшить рост таких важных в коммерческом отношении бактерий, как Е. coli, StreptomyceslividansrCorynebacteriumglutarnicumи Xanlhomonasmaltophilia, а также осуществлять в них экспрессию чужеродных генов. Интеграция чужеродной ДНК в хромосому хозяинаПри наличии вклетке плазмиды часть энергетических ресурсов расходуется на ее репликацию, транскрипцию и синтез белков, которые она кодирует. При этом, как правило, многокопийные плазмиды требуют больше энергии, чем малоколийные, и в результате часть клеток в процессе роста популяции утрачивает плазмиды. Клетки, лишившиеся своих плазмид, обычно растут быстрее тех, в которых они сохранились, и в конечном счете оказываются в культуре преобладающими. По прошествии нескольких генераций это отражается на количестве синтезируемого продукта клонированного гена. Разработано по крайней мере два подхода к решению этой проблемы. В лабораторных условиях для сохранения плазмид клетки выращивают в присутствии антибиотиков или метаболитов, обеспечивающих рост только тех клеток, в которых есть плазмида. Однако добавление антибиотиков и какихто других веществ в культуры, выращиваемые в больших объемах, или в промышленные ферментеры приводит к значительному удорожанию конечного продукта. Особенно важно, чтобы клонированные гены сохранялись, не утрачиваясь и не передаваясь другим микроорганизмам, в том случае, когда сконструированный микроорганизм предназначен для использования вне стен лаборатории. Он должен не только оставаться эффективным, но и быть экологически безопасным. Включение клонированной ДНК в хромосомную ДНК хозяйского организма позволяет обойтись без плазмид и избежать утраты плазмидных генов. При встраивании нужного гена в хромосомную ДНК хозяина нужно позаботиться о том, чтобы сайт интеграции не находился внутри гена, кодирующего важную клеточную функцию. Для этого чужеродный ген включают в заведомо несущественный сайт. Кроме того, для обеспечения эффективной экспрессии его помещают под контроль регулируемого промотора. Для интеграции в нужный сайт вводимый ген должен содержать нуклеотидную последовательность длиной не менее 50 нуклеотидов, сходную с таковой в хромосомной ДНК, в пределах которых и должен произойти физический обмен (рекомбинация) между двумя молекулами ДНК. Вкратце процесс интеграции состоит в следующем. 1. Идентификация подходящего сайта интеграции, т. е. сегмента хозяйской ДНК, последовательность которого может быть прервана без ущерба для функционирования клетки. 2. Выделение и клонирование всего хромосомного сайта интеграции или его части. 3. Встраивание нужного гена вместе с регулируемым промотором в клонированный сайт интеграции {рис. 6.15, А) или вблизи него (рис. 6.15, Б). 4. Перенос полученной генетической конструкции «хромосомный сайт интеграции/клонированный ген» в хозяйскую клетку в составе плазмиды, не способной к автономной репликации в клетках этого хозяина. 5. Отбор и сохранение тех хозяйских клеток, которые экспрессируют клонированный ген. Наследование клонированного гена возможно только в случае его интеграции в хромосому клеток хозяина. Если клетка трансформирована нереплицируюшейся плазмидой, несущей клонированный ген в середине клонированного фрагмента с хромосомным сайтом интеграции, то может произойти спаривание между гомологичными нуклеотидными последовательностями плазмиды и хозяйской ДНК (рис. 6.15, А) и далее интеграция в результате двойного кроссинговера, осуществляемого ферментами клетки-хозяина. Альтернативный вариант - интеграция всей плазмидной ДНК в хромосому хозяина в результате одиночного кроссинговера (на рисунке не показано). Интеграция всей плазмиды может произойти и в том случае, если клонированный 124 ГЛАВА 6
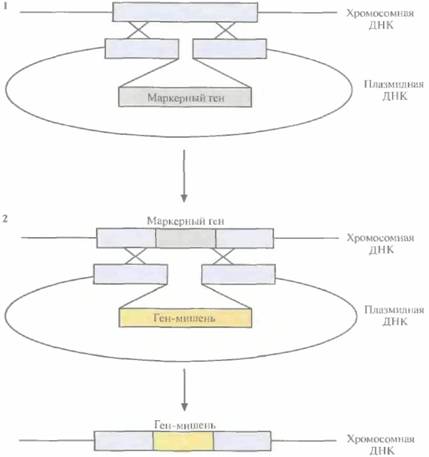
ген встроен вблизи клонированного хромосомного сайта интеграции. Для проверки эффективности интеграции клонированного гена использовали В. subtilis. Была сконструирована плазмида Е. соli, содержащая ген α-амилазы (фермента, участвуюшего в гидролизе крахмала) Bacillus amyloliquefaciens, встроенный в середину фрагмента ДНК из В. subtilis. Она была неспособна реплицироваться в этом микроорганизме, но ею можно было трансформировать клетки Б. sitbtilis. Обнаруженные трансформанты синтезировали α-амилазу, что свидетельствует об интеграции гена, кодирующего данный фермент, в хромосому В. subtilis, и о его функционировании. Отобранные рекомбинанты были устойчивы к ампициллину и хлорамфениколу. Поскольку оба гена устойчивости находились в плазмиде, было очевидно, что произошла одиночная рекомбинация, в результате которой вся плазмида включилась в хромосомную ДНК В. sitbtilis. Чтобы увеличить число копий гена α-амилазы, локализованных в хромосоме В. subtilis, ис- Оптимизация экспрессии генов, клонированных в прокариотических системах 125 ходные трансформанты выращивали в присутствии хлорамфеникола в высокой концентрации. В таких условиях выживали только те клетки, в которых происходила спонтанная дупликация интегрированной плазмиды. Клетки, отобранные по признаку устойчивости к хлорамфениколу, проверяли на активность α-амилазы (табл. 6.5). После такой процедуры были получены клетки, содержащие до 9 копий гена α-амилазы. Уровень ферментативной активности в клетках, содержащих α-амилазные гены в составе хромосомы, была гораздо выше, чем в том случае, когда эти гены находились в многокопийной плазмиде (от 20 до 40 копий на клетку) В. subtilts. В одном из исследований несколько копий чужеродного гена было встроено в разные заранее выбранные сайты в хромосоме В. sitbtilis, при этом для каждой из копий использовалась двух-этапная процедура (рис. 6.16), На первом этапе Таблица 6.5, Связь между числом копий гена α-амилазы и уровнем ее активности в клетках В. subtilis1)
1) По данным работы Kallio et al., I987. Appl. Micrcbiol. Biotechnol. 27: 64-71. выбирали селективный маркерный ген (например, ген устойчивости к какому-либо антибиотику) и встраивали его в середину вполне определенного, но несущественного фрагмента хромосомной ДНК В. subtilis в составе плазмид-
126 ГЛАВА 6 ного вектора, не способного к репликации в этом микроорганизме. Отбирали клетки, экспрессиру-ющие маркерный ген, т. е. клетки, у которых этот ген включился в хромосомную ДНК. На втором этапе ген-мишень с соответствующими сигналами инициации транскрипции и трансляции, введенный в середину такого же, как и выше, несущественного фрагмента хромосомной ДНК В. subtilisв составе плазмиды, включали в хромосомную ДНК с помощью рекомбинации, заменив им маркерный ген. Отбирали клетки, которые уже не экспрессировали маркерный ген, т. е. несли вместо него ген-мишень, интегрированный в хромосомную ДНК. Для интеграции других копий гена-мишени в хромосомную ДНК хозяйской клетки повторяли эту процедуру, используя другие несущественные области ДНК. Повышение эффективности секрецииСтабильность белков, кодируемых клонированными генами, зависит от их клеточной локализации. Например, рекомбинантный проинсулин оказывается примерно в 10 раз более стабильным, если он секретируется (экспортируется) в периплазму (пространство между плазматической и наружной мембранами), а не остается в цитоплазме. Кроме того, белки, секретируемые в периплазму или в среду, легче очистить. Обычно транспорт белков через клеточную мембрану обеспечивают N-концевые аминокислотные последовательности, называемые сигнальными пептидами (сигнальными последовательностями, лидерными пептидами). Иногда удается сделать белок секретируемым, присоединив к кодирующему его гену нуклеотидную последовательность, ответственную за синтез сигнального пептида. Однако простое наличие сигнального пептида не обеспечивает эффективной секреции. Кроме того, E. coli и другие грамотрицательные микроорганизмы обычно не могут секретировать белки в окружающую среду из-за наличия наружной мембраны. Есть по крайней мере два способа решения этой проблемы. Первый — использование грамположительных про- или эукариот, лишенных наружной мембраны, второй — создание грамотрицательных бактерий, способных секретировать белки в среду, с помощью генной инженерии. Если слияние гена-мишени с фрагментом ДНК, кодирующим сигнальный пептид, не приводит к эффективной секреции белкового продукта, приходится использовать другие стратегические приемы. Один из таких приемов, с успехом примененных в отношении ин-терлейкина-2, основывался на слиянии гена, кодирующего интерлейкин-2, с геном, кодирующим полноразмерный предшественник мальтозосвязывающего белка, а не только его сигнальную последовательность, и разделении этих генов сегментом ДНК, кодирующим сайт узнавания для фактора Ха. Когда такой химерный ген включили в плазмидный вектор и использовали его для трансформации E. coli, в периплазме хозяйской клетки обнаружили в большом количестве химерный белок. Обработав его фактором Ха, получили функциональный интерлейкин-2. По данным одной из работ, секреция многих гетерологичннх белков в E. coli зависит от уровня экспрессии соответствующих генов. Чужеродные белки, синтезируемые наиболее активно, не обязательно столь же активно секретируются. Иногда интенсивный синтез чужеродного белка вызывает перегрузку секреторного аппарата и его блокирование. Таким образом, если нужно, чтобы данный белок непременно секретировался, то можно попытаться понизить уровень экспрессии соответствующих генов. Некоторые грамотрицательные бактерии секретируют в среду белок, называемый бактериоцином. Он активирует фосфолипазу А, локализованную во внутренней мембране бактериальной клетки, в результате чего и внутренняя, и наружная мембраны становятся проницаемыми, и некоторые цито- и периплазматические белки высвобождаются в культуральную среду. Таким образом, можно встроить ген бактериоцина в плазмиду так, чтобы он находился под контролем сильного регулируемого промотора, трансформировать клетки E. coli этой плазмидой и сделать их проницаемыми. Если же E. coli уже несут ген бактериоцина, их можно трансформировать другой плазмидой, которая содержит ген нужного белка, сшитый с нуклеотидной последовательностью, кодирующей сигнальный пептид, Если оба гена находятся под контролем одного промотора, то их можно индуцировать одновре- Оптимизация экспрессии генов, клонированных в прокариотических системах 127 менно, и белок клонированного гена будет секретироваться в среду. Когда секретируемые чужеродные белки образуются в Е. coli в слишком большом количестве, очень часто процессинг претерпевают не все белки-предшественники: примерно половина секретированных белков сохраняет лидерную последовательность, а другая половина полностью процессируется с образованием зрелой формы. Это может быть связано с недостатком каких-то белков, участвующих в секреции. В такой ситуации, чтобы увеличить долю процесси-рованных белков, можно попытаться повысить уровень экспрессии генов, ответственных за синтез лимитирующих компонентов секретиру-юшей системы. Для проверки этого предположения были поставлены следующие эксперименты. Плазмиду, несущую гены prlA4 и secE, которые кодируют основные компоненты молекулярного механизма, ответственного за физическое перемещение белков через мембрану, ввели в клетки E. соli. После такого усиления секреторного аппарата хозяйской клетки доля ре-комбинантного белка (цитокина интерлейкина-6), секретируемая в плазмиду в зрелой форме, увеличилась с 50 до более чем 90%. Грибы Aspergilluscекретируют в среду большое количество ферментов и широко используются для их промышленного производства. В одной из работ осуществили слияние гена человеческого интерферона с геном сигнального пептида, ответственного за секрецию, и поместили эту конструкцию под контроль глюкоамилазного промотора Aspergitlusnidulans, индуцируемого крахмалом. После добавления последнего в среду с трансформированными клетками Aspergillusnidulansвыход секретируемого человеческого интерферона достиг 1 мг на 1 л, что эквивалентно примерно 5% всего секретируемого клеточного белка. Эта работа показывает, что стратегии модулирования генной экспрессии, разработанные для Е, соli, можно использовать и применительно к другим биологическим системам. Метаболическая перегрузкаВведение в клетку чужеродной ДНК и ее экспрессия часто приводят к нарушениям клеточного метаболизма. Эти нарушения весьма разнообразны и обусловлены давлением, которое оказывает чужеродная ДНК на все клеточные процессы. Метаболическая перегрузка может возникать по разным причинам. • Увеличение числа копий и/или размера плазмид и связанное с этим увеличение количества энергии, необходимого для их репликации и сохранения, • Недостаток растворенного кислорода в среде и невозможность обеспечения им и всех метаболических реакций, и процесса экспрессии плазмидных генов, • Гиперпродукция чужеродных белков, приводящая к истощению пула некоторых аминоацил-тРНК (или даже некоторых аминокислот) и/или энергетических запасов (в виде АТР и GTP). • Перегрузка системы экспорта и нарушение правильной локализации жизненно важных белков хозяйской клетки вследствие «перепроизводства" чужеродного белка, экспортируемого из цитоплазмы к клеточной мембране или в пери плазматическое пространство. • Наличие у организма-хозяина необычных метаболических свойств (например, высокая дыхательная активность у Azotobacterspp.), что делает его более чувствительным к различным воздействиям, чем обычные клетки. • Непосредственное влияние чужеродных белков на функционирование хозяйской клетки (например, превращение с их помощью важных незаменимых предшественников в неприемлемые, а иногда и токсичные соединения). Метаболическая перегрузка может привести к разнообразным изменениям в физиологии и функционировании хозяйской клетки. Одно из наиболее частых — снижение скорости роста клеток после введения чужеродной ДНК. Так, клетки, содержащие плазмиду, растут медленнее, чем нетрансформированные, не содержащие плазмид (табл. 6.6), что часто сопровождается утратой рекомбинантной плазмиды. Иногда метаболическая перегрузка приводит к тому, что под давлением отбора из плазмиды делегируется рекомбинантный ген или его часть. 128 ГЛАВА 6
Поскольку клеткам, растущим в условиях метаболической перегрузки, не хватает энергии для нормального функционирования, затрагиваются прежде всего такие энергоемкие метаболические процессы, как фиксация азота или синтез белков. Могут изменяться также размер и форма клеток, образовываться слишком много внеклеточного полисахарида, склеивающего клетки друг с другом и затрудняющего микрофильтрацию. Как следствие метаболической перегрузки, обусловленной образованием избыточного количества чужеродного белка и нехваткой питательных веществ или «строительных блоков» -аминокислот, может произойти запуск стрессовых механизмов, в частности инициироваться синтез клеточных протеиназ, под действием которых произойдет быстрая деградация рекомбинантного белка. Истощение пула аминокислот может стать результатом эффективной экспрессии не только клонированных генов-мишеней, но и генов самого вектора, кодирующих маркеры устойчивости к антибиотикам. Вероятность трансляционных ошибок для E. coli составляет 2·104—2·103 на 1 клетку за генерацию. Однако в условиях нехватки определенных аминоацил-тРНК, что часто случается при суперпродукции чужеродных белков, вероятность включения в белковую молекулу неправильной аминокислоты вместо недостающей сильно увеличивается. Кроме того, точность трансляции еше больше снижается из-за недостатка GTP, который является необходимым компонентом корректирующего аппарата. В одной из работ было показано, что в условиях гиперпродукции фактора роста эпидермиса мыши в клетках E. coli частота ошибочных включений аминокислот в рекомбинантный белок увеличивается в 10 раз. Это не позволяет использовать синтезируемый белок в качестве лекарственного средства, поскольку: 1) удельная активность и стабильность белка могут быть гораздо ниже ожидаемых; 2) наличие в молекуле «неправильных» аминокислот может вызвать нежелательную иммунологическую реакцию при введении такого белка в организм человека. К счастью, правильно спланировав эксперимент, можно минимизировать влияние метаболической перегрузки, оптимизировать выход рекомбинантного белка и повысить стабильность трансформированных хозяйских клеток. Например, нагрузку можно снизить, если использовать малокопийные плазмидные векторы. А еще лучше вообще отказаться от векторов и встроить чужеродную ДНК в хромосомную ДНК организма-хозяина. В этом случае не нужно заботиться об обеспечении стабильности плазмиды. Кроме того, клетке не приходится расходовать свои ресурсы на синтез ненужных продуктов, кодируемых маркерными генами устойчивости к антибиотикам. Синтез продуктов таких генов, входящих в состав плазмидных векторов наряду с генами-мишенями, является одной из основных причин метаболической перегрузки. Интеграция в хромосому особенно важна в тех случаях, когда используется сам рекомбинантный микроорганизм, а не синтезируемый им продукт. Уменьшению метаболической перегрузки помогает также применение сильных, но регулируемых промоторов, В такте случаях ферментацию проводят в две стадии. На первой из них, во время роста, промотор, контролирующий транскрипцию гена-мишени, выключен, а на второй, во время индукции, -включен. Оптимизация экспрессии генов, клонированных в прокариотических системах 129 Если частота использования кодонов у чужеродного гена отличается от таковой у организма-хозяина, то проблему нехватки специфических аминоацил-тРНК можно решить, синтезировав часть гена-мишени или даже весь ген с более близким к хозяйскому организму набором кодонов. Так, в одном из исследований было показано, что количество стрептавидина, образующегося при экспрессии синтетического гена с GС-содержанием 54%, было в 10 раз больше, чем при экспрессии «природного» гена с GC-содержанием 69%, Однако этот подход довольно сложен и может применяться лишь в редких случаях. Как это ни парадоксально, но один из способов увеличения количества чужеродного белка, синтезируемого рекомбинантным микроорганизмом, состоит в поддержании уровня экспрессии его гена на среднем уровне (так, чтобы на долю продукта приходилось примерно 5% суммарного клеточного белка), но зато в максимальном увеличении плотности культуры. Микробиологическая система с 5%-ным уровнем экспрессии чужеродного белка и низкой метаболической нагрузкой, в которой плотность может достигать 40 г/л (масса сухого вещества), оказывается более эффективной, чем система с 15%-ным уровнем экспрессии и плотностью 10 г/л. Достичь одновременно и высокого уровня синтеза чужеродного белка, и высокой плотности культуры часто не удается из-за накопления вредных побочных продуктов (в первую очередь ацетата), подавляющих рост клеток и синтез белка. Чтобы уменьшить накопление ацетата в богатой среде, не нарушая роста клеток, можно снизить скорость поглощения глюкозы, добавив в среду ее аналог, метил-α-гликозид. Альтернативный подход состоит в использовании клеток Е. coli, несущих мутацию в гене рtsG, который кодирует фермент II глюкозофосфотрансферазной системы. Максимальная плотность культуры Е. coliдикого типа составила примерно 10 г/л, а культуры E. coli с мутацией в гене ptsG — 15 г/л. Кроме того, уровень синтеза ß-лактамазы в мутантных клетках был на 25% выше (на 1 г массы сухого вещества), чем в клетках дикого типа, так что суммарное различие достигает примерно двукратной величины. Достичь аналогичного результата можно гораздо проще и быстрее, если использовать методы генной инженерии, а не мутагенез и отбор. Один из подходов состоял во введении в E. coli
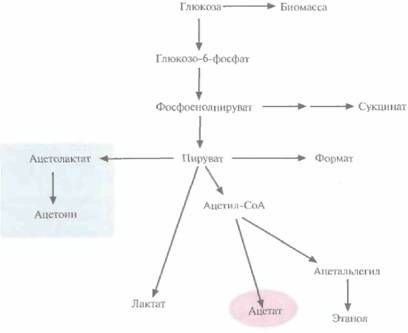
130 ГЛАВА 6 генов, кодирующих ацетолактатсинтазу. Этот фермент катализирует образование ацетолактата из пирувата, что приводит к уменьшению количества образующегося ацетата (рис. 6.17). Гены ацетолактатсинтазы вводят в клетки в составе одной плазмиды, а гены-мишени — в составе другой, из другой группы несовместимости. Трансформированные клетки синтезируют гораздо меньше ацетата, чем нетрансформированные; вместо него образуется ацетоин, соединение примерно в 50 раз менее токсичное, чем ацетат. ЗАКЛЮЧЕНИЕЧтобы получить какой-то белковый продукт, необходимо обеспечить правильную транскрипцию кодирующего его гена и трансляцию соответствующей мРНК. Для инициации транскрипции в нужном сайте необходим промотор, а для ее остановки - терминирующий кодон. Клонированный ген часто бывает лишен таких сигнальных последовательностей, и для его экспрессии в прокариотической клетке-хозяине нужно обеспечить и то, и другое. Кроме того, поскольку для решения большинства биотехнологических задач белок должен образовываться в больших количествах, необходимо использовать промотор, который позволял бы получить высокий уровень транскрипции (сильный промотор) и распознавался РНК-полимеразой хозяйской клетки. Постоянная транскрипция клонированного гена истощает энергетические ресурсы хозяйской клетки, поэтому нужно использовать промоторы, работу которых можно регулировать либо с помощью специфических низкомолекулярных соединений, либо изменением температуры. Эффективность синтеза белка зависит от наличия в его мРНК специфических нуклеотидных последовательностей. Чтобы предотвратить разрушение белкового продукта или обеспечить его секрецию, клонированные гены, которые кодируют этот белок, подвергают направленным изменениям. Это может быть присоединение сайта связывания рибосомы перед сайтом инициации транскрипции (который в свою очередь тоже бывает нужно присоединить) или добавление к концу клонированного гена терминирующего кодона, который обеспечивал бы остановку трансляции. Если нужно, чтобы белок секретировался, то перед клонированным геном необходимо встроить сигнальную последовательность, рамка считывания которой согласуется с таковой гена-мишени. Еще одна проблема — невысокая стабильность белков, кодируемых клонированными генами, Рекомбинантный белок может расщепляться про-теиназами хозяйской клетки. Чтобы избежать этого, можно изменить клонированный ген таким образом, чтобы на N-конце белковой молекулы оказались одна или несколько дополнительных аминокислот. В такой форме рекомбинантный белок уже не подвергается столь быстрой деградации. Кроме того, «лишние» аминокислоты иногда помогают в последующей очистке химерного белка, например с помощью иммуноаффинной хроматографии на колонке. При этом место соединения компонент подбирается так, чтобы по нему можно было расщепить молекулу (химическим или ферментативным путем) и получить эти компоненты в чистом виде. Большинство микроорганизмов, с помощью которых получают белковые продукты, растут только в присутствии кислорода. Последний плохо растворяется в воде, и при интенсивном росте его запасы быстро истощаются. Чтобы обойти эту трудность, использовали два пути: 1) применяли штаммы, неспособные синтезировать некоторые протеолитические ферменты; 2) включали в геном хозяйских клеток гены, кодирующие гемоглобин Vitreoscillasp., который связывает кислород окружающей среды и повышает его концентрацию в клетке. С увеличением числа копий клонированного гена увеличивается количество синтезируемого продукта. Однако при переходе к крупномасштабному производству конструкция «плазмида/клонированная ДНК» очень часто утрачивается. Чтобы избежать этого, разработали способы интеграции клонированного гена в хромосому организма-хозяина. В этом случае ген остается в клетке как часть хозяйской ДНК. Введение в геном организма-хозяина и экспрессия чужеродной ДНК часто приводят к нарушению его метаболизма и нормального функционирования — так называемой метаболической Оптимизация экспрессии генов, клонированных в прокариотических системах 131 перегрузке. Разработан целый ряд способов, позволяющих минимизировать этот эффект и одновременно оптимизировать выход белка-мишени и стабильность трансформированных клеток. Системы экспрессии весьма разнообразны, и исследователям приходится каждый раз подбирать условия, наиболее подходящие для получения того или иного белка в том или ином организме-хозяине. И все же, несмотря на различия в деталях, для создания самых разных систем экспрессии используются одни и те же основные приемы. ЛИТЕРАТУРААшапл E., J. Brosius. 1985. "ATG vectors" for regulated high-level expression of clones genes in Escherichia coll. Gene A»: 183-190. Aristidou A. A., K. Y. San, G. N. Bennett. 1995. Metabolic engineering of Escherichia coli to enhance recombinant protein production through acetate reduction. Biotechnol Prog. II:475—478. BachniairA., D, Finley, A. Varshavsky. 1986. In vivo half-life of a protein in a function of its arnino-termimit residue. Science 234: 179—186. Bagdasarian M. M., E. Amann, R. Lurz, B. Ruckert, M. Bagdasarian. 1983. Activity of the hybrid Op-lac (tac) promoter of Escherichia coli in Pseudomonas putida. Construction of broad-host-range, controlled-expression vectors, Gene 26: 273-282. Baker R. T., A. Varshavsky. 1991. Inhibition of the N-end rule pathway in living cells. Proc. Naît. Acad. Set. Î/&488: 1090-1094. Chaitan K., J. E. Curtis, J. De Modena, U. Rinas, J. E. Bailey. 1990. Expression of intracellular hemoglobin improves protein synthesis in oxygen-limited Escherichia coli. Bio/Technology 8: 849-853. Chou С. H., G. N. Bennett, K. V. San. 1994. Effect of modified glucose uptake using genetic engineering techniques on high-level recombinant protein production in Escherichia coli dense cultures. Biotechnol. Bioeng. 44: 952-960. deBoer H. A., L J. Cnmstnck, M. Yasser. 1983. The tac promoter a functional hybrid derived from the trp and lac promoters. Proc. Nafl. Acad. Set. USA 80: 21-25. Donovan R. S., C. W. Robinson, B. R. Click. 1996. Optimizing inducer and culture conditions for expression of foreign proteins under the control of the lac promoter. /. Ind. Microbiol. 16: 145-154. Ernst J. F. 1988. Codon usage and gene expression. Trends Biotechnol. 6: 196-199. Friesen J. D., G. An. 1983. Expression vehicles used in recombinant DNA technology. Biotechnol. Adv. 1: 205-227. Geisow M. J. 1991. Both bane and blessing-inclusion bodies. Trends Biotechnol. 9: 368-369. Gentz R., A. Langner, A. C. Y. Chang, S. N. Cohen, fL Bujard. 1981. Cloning and analysis of strong promoters is made possible by the downstream placement of a RNA termination signal, Proc. Natl. Acad. Sei. USA 78: 4936-4940. Click B. R. 1995. Metabolic load and heterologous gene expression. Biotechnol. Adv. 13: 247—261, Click B. R., G. K. Whitney. 1987. Factors affecting the expression of foreign proteins in Escherichia coli. J. Ind. Microbiol. I: 277-282. Goldstein M. A., R. H. Doi. 1995. Prokaryotic promoters in biotechnology, In p. 105—128. M. R. El-Gewely (ed.), Biotechnology Annual Review, vol. 1. Elsevier Science B. V., Amsterdam, The Nertherlands. Gwynne D. I., F. P. Buxton, S. A. Williams, S. Garven, R. W. Davies. 1987. Genetically engineered secretion of active human interferon and a bacterial endoglucanase from Aspergillus nidu-ians. Bio/Technology 5: 713-719. Halfmann G., H. Brailly, A. Bernadac, F. A. Mon-tero-Julian, C. Lazdimski, D. Baty. 1993. Targeting of interleukin-2 to the periplasm of Escherichia coli. J. Gen. Microbiol. 139: 2465-2473. Hartley J. L., T. J. Gregori. 1981. Cloning multiple copies of a DNA gene. Geve 13: 347-353. Hockney R. C. 1994. Recent developments in heterologous protein production in Escherichia coli. Trends Biotechnol. 12: 456-463. Hopp T. P., K. S. Pricket t, V. L. Price, R. T. Libby, C. J. March, D. P. Cerretti, D. L. Urdal, P. J. Conlon. 1988. A short polypeptide marker sequence useful for recombinant protein identification and purification. Bio/Technology 6: 1204-1210. Hsiung H. M., A. Cantrell, J. Luirink, B. Oudega, A. J. Veros, G. W. Becker 1989. Use of bacteri- 132 ГЛАВА 6 ocin release protein in E. coll for excretion of human growth hormone into the culture medium. Bio/Technology 7: 267-271. Jay G., G. Khoury, A. K. Seth, E. Jay. 1981. Construction of a general vector for efficient expression of mammalian proteins to bacteria: use of a synthetic ribosome binding site. Proc. Natl. Acad. Sei. USA 78: 5543-5548. Jespers L. S., J. H. Messens, A. De Keyset. D. Lee kin »tit, 1. Van den Brande, Y. (i. (Îansemaiis. M. J. Lauwereys, G. P. Vlasuk, P. E. Stanssens. 1995. Surface expression and ligand-based sélection of cDNAs fused to filamentous phage gene VI. Bio/Technology 13: 378-382. Kaffio P., A. Palva, I. Palva. 1987. Enhancement of α-amylase production by integrating and amplifying the α-amylase gene of Bacillus amylotique-faciens in the genome of Bacillus subtilis. Appt. Microbiol. Biotechnol 27: 64-71. Kiel J. A. K. W., A. M. ten Berge, P. Borger, G. Venema. 1995. A general method for the consecutive integration of single copies of a heterol-ogous gene at multiple locations in the Bacillus subtilis chromosome by replacement recombination. Appf. Environ. Microbiol. 61: 4244—4250. Kolata G. 1986. New rule proposed for protein degradation. Science234: 151—153. Kolowsky K. S., J. G. K. Williams, A. A. Szalay. 1984. Length of foreign DNA in chimeric plas-mids determines the efficiency of its integration into the chromosome of the cyanobacterium Synechococcus R2. Gene 27: 289-299. Kozlowski M., A. VanBrunschot, G. Nash, R. W. Davies. 1988. A novel vector allowing the expression of genes in a wide range of gram-negative bacteria. Gene 70: 199-204. Labes M., A. Puhler, R. Simon. 1990. A new family of RSFlOlO-derived expression and /йс-fusion broad-host-range vectors for gram-negative bacteria. Gene 89: 37-46. Lee N., J. Cozzitorto, N. Wainwright, D. Testa. 1984. Cloning with tandem gene system for high level gene expression. Nucleic Acids Res. 12: 6797-6812. Leemans R., E. Remaut, W. Fiers. 1987. Broad-host-range expression vector based on the pLpromoter of соliphage 1: regulated synthesis of human interleukin 2 in Erwinia and Serratia species. J. Bacterioi. 169: 1899-1904. Little M., F. Breitling, B. Micheel, S. Dübel. 1994. Surface display of antibodies, Biotechnol. Adv. 12: 539-555. Liu S. C., D. A. Webster, M. L. Wei, В. С. Stark. 1996. Genetic engineering to contain the Vitreoscilla hemoglobin gene enhances degradation of be n zoic acid by Xanthomonas maltophilia. Biotechnol. Bioeng. 49: 101-105. Looman A. C., J. Bodlaender, M. de Gruyter, A. Vogelaar, P. H. van Knippenberg. 1986. Secondary structure as primary determinant of the efficiency of ribosomal binding sites in Escherichia coii. Nucleic Acids Res. 14: 5481-5497. Magnolo S. K., D. L. Leenutaphong, J. A. DeMu-dena, J. E. Curtis, J. E. Bailey, J. L. Gala//«, D. E. Hughes. 1991. Actinorhodin production by Streptomyces coeticoior and growth of Streptomyces lividans are improved by the expression of a bacterial hemoglobin. Bio/Technology 9: 473-476. Meerman H. J., G. Georgiou. 1994. Construction and characterization of a set of E, coli strains deficient in all known loci affecting the prote-olytic stability of secreted recombinant proteins. Bio/Technology 12: 1107-1110. Miescliendahl M., B. Miiller-HUl. 1985. F'-coded, temperature -sensitive λ cI857 represser gene for each construction and regulation of λ promoter-dependent expression systems. J. Bacterioi. 164: 1366-1369. Mieschendahl M., T. Pétris, U. Hanggi. 1986. A novel prophage independent trp regulated XpL expression system. Bio/Technology 4: 802-808. Murby M., M. Üblen, S. Stähl. 1996. Upstream strategies to minimize proteolytic degradation upon recombinant production in Escherichia coii. Protein Expr. Purif. 7: 129-136. Nagai K., H. C. Thogersen, 1984, Construction of ß-globin by sequence-specific proteolysis of a hybrid protein produced in Escherichia coii. Nature 309:810-812. Nygren P. Λ., S. Stahl, M. Uhlén. 1994. Engineering proteins to facilitate bioprocessing. Trends Biotechnol. 12; 184-188. O'Neil К. Т., R H. Hoess. 1995. Phage display: protein engineering by directed evolution. Curr, Opin. Struct. Biol. 5: 443-449. Оптимизация экспрессии генов, клонированных в прокариотических системах 133 Perez-Perez J., G. Marquez, J. L. Barbero, J. Gutierrez. 1994. Increasing the efficiency of protein export in Escherichia con". Bio/Technology 12: 178-180. Remaut E., H. Tsao, W. Fiers. 1983. Improved plas-mid vectors with thermoinducible expression and temperature-regulated runaway regulation. Gene 22: 103-113. Rogers S., R. Wells, M. Rechsteiner. 1986. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234: 364-368. Sander F. C., R. A. Fachini, D. £. Hughes, J. L. Galazzo, J. E. Bailey. 1994. Expression of Vitreoscilla hemoglobin in Corynebacterium glu-tamicum increases final concentration and yield of L-Jysine, p. 607-610, /« L. Alberghina, L. Frontali. P. Sensi ied.). Proceedings of the 6th European Congress on Biotechnology. Eisevier Science B.V., Amsterdam, The Netherlands. Sassenfeld H. M., 1990. Engineering proteins for purification. Trends Biotechnol. 8: 88—93. Simmons L. C., D. G. Yansura. 1996. Translational level is a critical factor for the secretion of heterol-ogous proteins in Escherichia coli. Nat. Biotechnol. 14: 629-634. Sung W. L., F. L. Yao, D. M. Zahab, S. A. Narang. 1986. Short synthetic oligodeoxyribonucleotide leader sequences enhance accumulation of human proinsulin synthesized in Escherichia coli. Proc. Nail. AcaO. Sei. USA 83: 561-565. Talmadge K., W. Gilbert. 1982. Cellular location affects protein stability in Escherichia coli. Proc. Nail. AcaO. Sei. USA 79: 1830-1833. Taylor W. M., P. J. Hagerman. 1987. A general method for cloning DNA fragments in multiple copies. Gene 53: 139-144. Tobias J. W., T. E. Schrader, G. Rocap, A. Varshavsky. 1991. The N-end rule in bacteria. Science 254: 1374-1377. Tsuchiya M., Y. Morinaga. 1988. Genetic control systems of Escherichia coli can confer inducible expression of cloned genes in coryneform bacteria. Bio/Technology 6: 428-430. Weinstock G. M., C. A. Rhys, M. L. Berman, B. Hampar, D. Jackson, T. J. Silhavy, J. Weisemann, M. Zweig. 1983. Open reading frame expression vectors: a general method for antigen production in Escherichia coli using pro- tein fusion to ß-galactosidase. Proc. Natl, Acad. Sei. USA 80: 4432-4436. Wilcox G., G. M. Studnicka. 1988. Expression of foreign proteins in microorganisms. Biotechnol. Appl. Biochem. 10: 500-509. Wilkinson D. L., R. G. Harrison, 1991. Predicting the solubility of recombinant proteins in Escherichia coli. Bio/Technology 9: 443—448. Williams J. G. K., A. A. S/alay. 1983. Stable integration of foreign DNA into the chromosome of the cyanobacterium Synechococcus R2. Gene 24: 37-51. Wong R. S. Y., R. A. Wirtz, R, E. W. Hancock. 1995. Pseudomonas aeruginosa outer membrane protein OprF as an expression vector for foreign epitopes: the effects of positioning and length on the anti-genicity of the epitope. Gene158: 55—60. КОНТРОЛЬНЫЕ ВОПРОСЫ1. Какими способами можно влиять на экспрессию генов, клонированных в прокарио-тических организмах? 2. Что такое ген lacIqи как его используют? 3. Почему плазмидный вектор с максимально сильным промотором не всегда является наилучшим экспрессирующим вектором? 4. Что такое tac-промотор и как осуществляется его регуляция? 5. Промотор pLфага λ, способного инфицировать только E. coli, тем не менее иногда используют как составную часть экспрессиру-ющего вектора с широким кругом хозяев. Как «приспособить» рL-промотор для инициации транскрипции в других организмах? 6. Иногда стратегия синтеза белка-мишени включает получение этого белка в составе химерного продукта. В чем преимущество такого подхода? Как создают химерный белок? 7. Что такое тельца включения и как избежать их образования? 8. В чем преимущество локализации чужеродных белков на поверхности клеток? Какие стратегии используются для того, чтобы сделать белки секретируемыми? 9. Как встроить в одну плазмиду несколько копий гена? 134 ГЛАВА 6 10. Как решить проблему обеспечения кислородом клеток Е. соli, синтезирующих в большом количестве чужеродный белок? 11. Последовательность-мишень может быть встроена в хромосомную ДНК двумя способами: 1) сама по себе; 2) в составе плазмиды, которая несет эту последовательность. Как происходит каждое из этих событий? Какие преимущества или недостатки имеет интеграция плазмидного вектора в хозяйскую ДНК? 12. Что такое метаболические перегрузки и какова их причина? 13. Предложите несколько способов снижения метаболической перегрузки E. coli, синтезирующих в большом количестве рекомбинантный белок. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||