Струков Патологическая анатомия. Литература для студентов медицинских институтов А. И. Струков В. В. Серов Патологическая анатомия Издание второе
 Скачать 16.97 Mb. Скачать 16.97 Mb.
|

|
Механизм развития гидропической дистрофии сложен и отражает наруше- ния водно-электролитного и белкового обмена, ведущие к изменению кол- лоидно-осмотического давления в клетке. Большую роль играет наруше- ние проницаемости мембран клетки, сопровождающееся их распа- дом. Это ведет к закислению цитоплазмы, активации гидролитиче- ских ферментов лизосом, которые разрывают внутримолекулярные связи с присоединением воды. 22 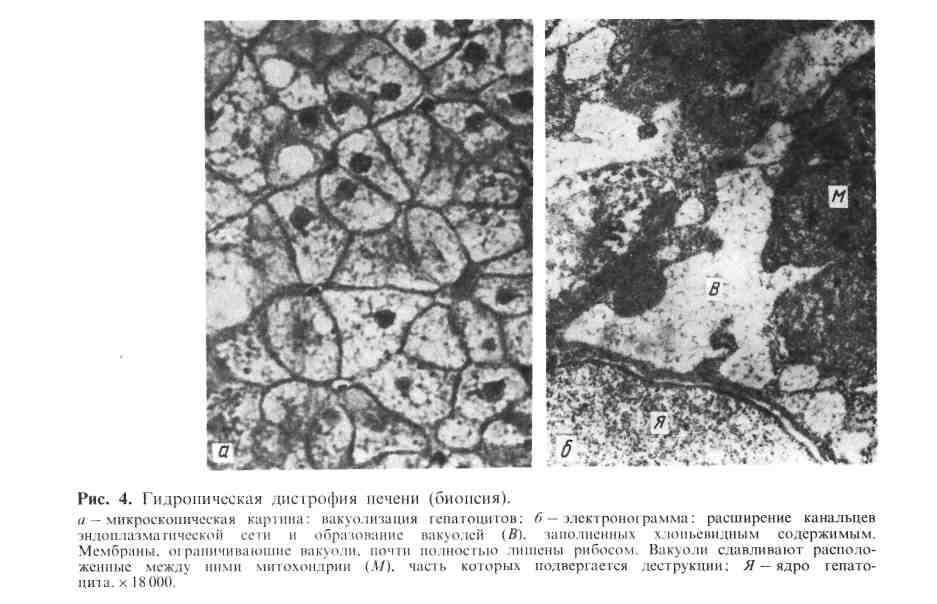 Исход гидропической дистрофии, как правило, неблагоприятный, так как она нередко переходит в баллонную, для которой характерно разрушение жизненно важных структур клетки. Функция органов и тканей при гидропической дистрофии резко снижена. Роговая дистрофия Роговая дистрофия, или патологическое ороговение, ха- рактеризуется избыточным образованием рогового вещества в ороговеваю- щем эпителии (гиперкератоз, ихтиоз) или образованием рогового ве- щества там, где в норме его не бывает (патологическое ороговение на слизистых оболочках, или лейкоплакия; в эпителиальных опухолях, на- пример образование «раковых жемчужин» в плоскоклеточном раке). Процесс может быть местным или распространенным. Причины роговой дистрофии разнообразны: нарушение развития кожи, хроническое воспаление, вирусные инфекции, авитаминозы и др_ Исход может быть двояким: устранение вызывающей причины в начале процесса может привести к восстановлению ткани, однако в далеко зашедших случаях наступает гибель клеток. Значение роговой дистрофии определяется ее степенью, распространен- ностью и длительностью. Длительно существующий местный очаг патологи- ческого ороговения слизистой оболочки (лейкоплакия) может явиться источ- ником развития раковой опухоли. Врожденный ихтиоз резкой степени, как правило, несовместим с жизнью. К группе паренхиматозных диспротеинозов примыкает ряд дистрофий, в основе которых лежат нарушения внутриклеточного метаболизма ряда ами- нокислот в результате наследственной недостаточности метаболизирующих 23 их ферментов, т. е. в результате наследственной ферментопатии. Эти дистрофии относятся к так называемым болезням накопления. Наиболее яркими примерами наследственных дистрофий, связанных с на- рушением внутриклеточного метаболизма аминокислот, являются ц и с т и - ноз, тирозиноз, фенилпировиноградная олигофрения (фе- нилкетонурия). Их характеристика представлена в табл. 1. Таблица 1
Паренхиматозные жировые дистрофии (липидозы) В цитоплазме клеток содержатся в основном липиды, которые образуют с белками сложные лабильные жиробелковые (липопротеидные) комплексы. Эти комплексы составляют основу мембран клетки. Липиды вместе с белками являются составной частью и клеточных ультраструктур. Помимо липопро- теидов, в цитоплазме встречаются и нейтральные жиры, которые пред- ставляют собой сложные эфиры глицерина и жирных кислот. В клетках жиры обнаруживаются в виде капель и зерен, которые не растворяются в воде (в отличие от гликогена) и в уксусной кислоте (в отличие от белков), но растворимы в спиртах, эфирах, кислоте, хлороформе. Поэтому для выявления жиров используют срезы нефиксиро- ванных замороженных или фиксированных в формалине тканей. Гистохимически жиры выя- вляются с помощью ряда методов: судан III и шарлах окрашивают их в красный цвет, Судан IV и осмиевая кислота — в черный, сульфат нильского голубого окрашивает жирные кислоты в темно-синий цвет, а нейтральные жиры — в красный. Следует отметить, что липиды, входящие в состав липопротеиДных комплексов, не обнаруживаются при гистохимическом исследовании. С помощью поляризационного микроскопа можно дифференцировать изотропные, напри- мер нейтральные, жиры и анизотропные, например холестерин и его эфиры, дающие характер- ное двойное лучепреломление. Нарушения обмена цитоплазматических жиров могут проявляться в увели- чении содержания жиров в клетках, где они обнаруживаются и в норме, в по- явлении жиров там, где они обычно не встречаются, и в образовании жиров необычного химического состава (А. И. Абрикосов). Обычно в клетках накап- ливаются нейтральные жиры. Паренхиматозная жировая дистрофия встречается наиболее часто там же, где и белковая,—в миокарде, печени, почках. В миокарде жировая дистрофия характеризуется появлением в мышечных клетках мельчайших жировых капель (пылевидное ожире- н и е). При нарастании изменений эти капли (мелкокапельное ожире- ние) полностью замещают цитоплазму (рис. 5). Большинство митохондрий при этом распадается, поперечная исчерченность волокон исчезает. Процесс имеет очаговый характер и наблюдается в группах мышечных клеток, распо- ложенных по ходу венозного колена капилляров и мелких вен. Внешний вид сердца зависит от степени жировой дистрофии. Если процесс выражен слабо, его можно распознать лишь под микроскопом, применяя спе- 24 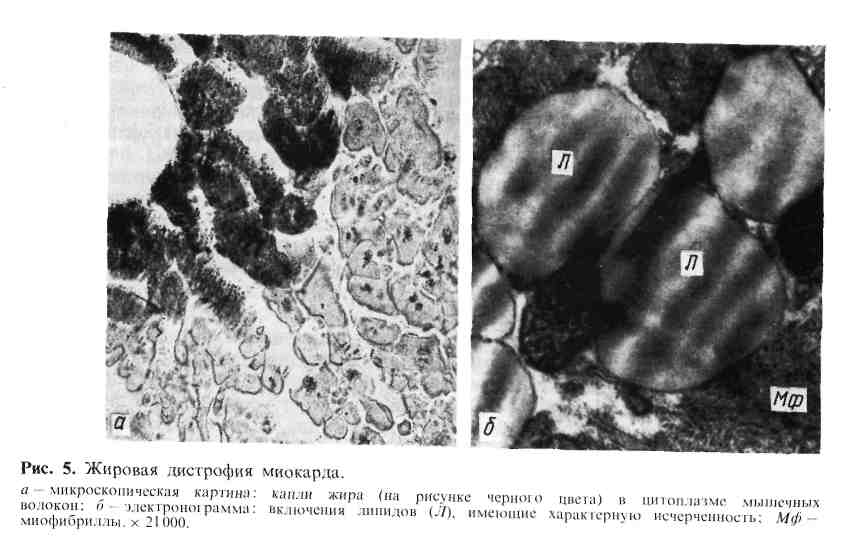 циальные окраски на жиры; если он выражен сильно, сердце выглядит увели- ченным в объеме, камеры его растянуты, оно обладает дряблой консистен- цией, миокард на разрезе тусклый, глинисто-желтый. Со стороны эндокарда видна желто-белая исчерченность (рис. Ь), особенно хорошо выраженная в со- сочковых мышцах и трабекулах желудочков сердца («тигровое сердце»). Эта исчерченность миокарда связана с очаговым характером дистрофии, преиму- щественным поражением мышечных клеток вокруг венул и вен. 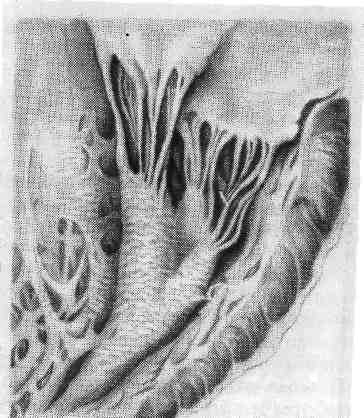 Впеченио жировой дистрофии (ожирении) говорят в тех случаях, когда содержание жиров в гепатоцитах резко увеличивается и меняется их состав. В клетках печени вначале появляются гранулы липидов (пылевидное ожирение), затем мелкие капли их (мелкокапельное ожирение), ко- торые в дальнейшем сливаются в круп- ные капли (крупнокапельное ожи- рение) или в одну жировую вакуоль, которая заполняет всю цитоплазму и отодвигает ядро на периферию. Изменен- ные таким образом печеночные клетки напоминают жировые. Чаще отложение жиров в печени начинается на периферии, реже— в центре долек; при значительно выраженной дистрофии ожирение клеток печени имеет диффузный характер. Внешний вид печени достаточно харак- терен: она увеличена, дряблая, охряно- Рис. 6. Жировая дистрофия миокарда («тигровое сердце»). Под эндокардом желто-белые полоски, соответствующие участкам включений липидов в мышечных клетках. 25 желтого или желто-коричневого цвета. При разрезе на лезвии ножа и по- верхности разреза виден налет жира. В п о ч к а х при жировой дистрофии жиры появляются в эпителии прокси- мальных и дистальных канальцев (рис. 7, см. на цветн. вкл.). Обычно это ней- тральные жиры, фосфолипиды или холестерин, который обнаруживают не только в эпителии канальцев, но и в строме. Нейтральные жиры в эпителии узкого сегмента и собирательных трубок встречаются как физиологическое явление. Внешний вид почек: они увеличены, дряблые (при сочетании с амилоидо- зом плотные), корковое вещество набухшее, серое с желтым крапом, за- метным на поверхности и разрезе. Причины жировой дистрофии разнообразны. Чаще всего она связана с кис- лородным голоданием (тканевой гипоксией), поэтому жировая дистрофия так часто встречается при заболеваниях сердечно-сосудистой системы, хрониче- ских заболеваниях легких, анемиях, хроническом алкоголизме и т. д. В усло- виях гипоксии страдают в первую очередь отделы органа, находящиеся в функциональном напряжении. Так, при пороке сердца жировая дистрофия миокарда возникает прежде всего в отделах сердца, подвергшихся компенса- торной гипертрофии. В связи с этим жировая дистрофия миокарда рассматри- вается как морфологический эквивалент декомпенсации сердца. Жировая дистрофия развивается при многих инфекциях (дифтерия, тубер- кулез, сепсис) и интоксикациях (фосфор, мышьяк, хлороформ), ведущих к на- рушениям обмена (диспротеиноз, гипопротеинемия, гиперхолестеринемия). В ряде случаев причинами могут быть авитаминозы и одностороннее (с недо- статочным содержанием белков) питание, сопровождающееся дефицитом фер- ментов и липотропных факторов, которые необходимы для нормального жи- рового обмена клетки. Такова, например, алипотропная жировая дистрофия печени, наблюдающаяся при белковом голодании. Механизм появления жира в клетке при жировой дистрофии может быть связан с декомпозицией, инфильтрацией или трансформа- цией и извращенным синтезом. Значение этих механизмов в разви- тии жировой дистрофии разных органов и тканей неравнозначно, что зависит от причины, вызвавшей дистрофию, и структурно-функциональных особенно- стей органа. Развитие жировой дистрофии миокарда связывают с тремя механизма- ми: повышенным поступлением жирных, кислот в кардиомиоциты, наруше- нием обмена жиров в этих клетках и распадом липопротеидных комплексов внутриклеточных структур. Чаще всего эти механизмы реализуются путем ин- фильтрации и декомпозиции (фанероза) при энергетическом дефиците миокар- да, связанном с гипоксией и интоксикацией. При этом основное значение де- композиции не в высвобождении липидов из липопротеидных комплексов клеточных мембран, а в деструкции митохондрий, что ведет к нарушению окисления жирных кислот в клетке. Жировая дистрофия печени, в развитии которой могут участвовать все морфогенетические механизмы встречается при: 1) чрезмерном поступлении в гепатоциты жирных кислот или повышенном синтезе их этими клетками; 2) воздействии токсических веществ, блокирующих окисление жирных кислот и синтез липопротеидов в гепатоцитах; 3) недостаточном поступлении в пече- ночные клетки аминокислот, необходимых для синтеза фосфолипидов и липо- протеидов. Из этого следует, что жировая дистрофия печени развивается при липопротеидемии (алкоголизм, сахарный диабет, общее ожирение, гормо- нальные расстройства), гепатотропных интоксикациях (этанол, фосфор, хло- роформ и др.), нарушениях питания (недостаток белка в пище, авитаминозы, болезни пищеварительной системы). 26 Жировая дистрофия почек чаще имеет резорбтивный характер, т. е. свя- зана с инфильтрацией эпителия почечных канальцев жиром при липемии и ги- перхолестеринемии (нефротический синдром), что ведет к гибели нефроцитов. Исход жировой дистрофии зависит от ее степени. Если она не сопрово- ждается грубым поломом клеточных структур, то, как правило, оказывается обратимой. Глубокое нарушение обмена клеточных жиров в большинстве слу- чаев заканчивается гибелью клетки. Функциональное значение жировой дистрофии очень велико; деятельность органов при ней резко нарушается, а в ряде случаев и выпадает. Группу наследственных липидозов составляют так называемые си- стемные липидозы, возникающие вследствие наследственного дефицита ферментов, участвующих в метаболизме определенных липидов. Поэтому си- стемные липидозы относят к наследственным ферментопатиям, или болезням накопления, поскольку дефицит фермента определяет накопление субстрата, т. е. липидов, в клетках. В зависимости от вида накапливающихся в клетках липидов различают: цереброзидлипидоз, или глюкозилцерамидлипидоз (болезнь Гоше), сфингомиелинлипидоз (болезнь Ниманна — Пика), ганглиозид- л и п и д о з (болезнь Тея — Сакса, или амавротическая идиотия), генерали- зованный ганглиозидоз (болезнь Нормана — Ландинга) и др. Чаще всего липиды накапливаются в печени, селезенке, костном мозге, центральной нервной системе (ЦНС), нервных сплетениях. При этом появляются харак- терные для того или иного вида липидоза клетки (клетки Гоше, клетки Пика), что имеет диагностическое значение при изучении биоптатов (табл. 2). Таблица 2 Системные липидозы (наследственные ферментопатии, болезни накопления, лизосомные болезни)
Многие ферменты, дефицит которых определяет развитие системных липи- дозов, относятся, как видно из табл. 2, к лизосомным. На этом основании ряд липидозов рассматривают как «лизосомные болезни». | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||