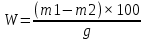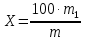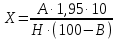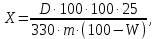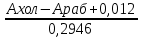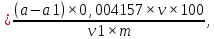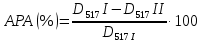ОТЧЕТ О ПРЕДДИПЛОМНОЙ ПРАКТИКЕ ФГБОУ ВО Красноярский ГАУ Разработка рыбо-растительных изделий для предприятий быстрого питания. Отчет по преддипломной практике (2). Отчет о преддипломной практике фгбоу во красноярский гау место прохождения практики
 Скачать 0.53 Mb. Скачать 0.53 Mb.
|

1.2 Методы исследования сырья и готовых рыборастительныхизделий1.2.1 Методы исследования растительного сырья – репы BrassicaRapaL.Для исследования химического состава репу мыли, очищали от кожуры, измельчали на блендере, сушили в проветриваемом помещении при температуре 20-22°С, с помощью механической мельницы получали более тонкий помол гранул исходного сырья - до 0,05-0,1 см. Для определения химического состава репы использованы классические химические (гравиметрия, титриметрия) и физико-химические методы анализа: рефрактометрия, УФ -и видимая спектроскопия. Ниже представлены методики определения содержания некоторых витаминов, редуцирующих сахаров, дубильных веществ, глюкорафанина в составе репы. Определение влажности Массовую долю воды в репе определяли путем высушивания навески в термошкафу при температуре 105° С до постоянного веса. Для количественного анализа содержания влаги использовали стеклянные бюксы с крышками, у которых заранее определена постоянная масса. С этой целью чистый бюкс ставили в термошкаф на 30 мин при температуре 105° С, затем помещали его в эксикатор с притертой крышкой для охлаждения и затем взвешивали. Операцию повторяли 3 раза до получения воспроизводимых данных В чистый и сухой бюкс помещали навеску 5-10 г анализируемого вещества. Бюкс с сырьем без крышки помещали в термошкаф. Высушивание проводили в течение 4–5 ч, далее процедуру повторяли – помещали в эксикатор, охлаждали, взвешивали и аналогично доводили до постоянной массы с использованием аналитических весов. Разница между параллельными результатами не должна превышать более 0,0001 г. Содержание влаги определяли по формуле:
где: W – массовая доля, %; g - навеска анализируемого сырья, г; m1 – масса бюкса с навеской до высушивания, г; m2 – масса бюкса с навеской после высушивания, г. Исследование зольности растительного сырья Для определения содержания минеральных веществ (зольности) в растительном сырье взвешивали точные навески проб (2-3 г) и обугливали сначала при температуре 200-250ºС в течение 2 часов и далее поднимали температуру до 500ºС и выдерживали еще 2 часа. Навескам давали остыть в муфельной печи. После остывания навесок их выдерживания в эксикаторе с CaCl2 не менее 1 часа, пробы взвешивали на аналитических весах. Расчет содержания минеральных веществ (зольности), в % проводили по формуле:
где m – масса абсолютно сухого исследуемого материала, г; m1 – масса золы, г Определение содержания витаминов С и РР Анализ сырья на витамины проводили фотометрическим методом на спектрофотометре СФ-46 в соответствии с ГОСТ 24556-89. Метод основан на выделении аскорбиновой кислоты из пробы метафосфорной кислотой, последующем восстановлении кислотой 2,6-дихлорфенол-индофенолята натрия, экстрагировании продукта восстановления ксилолом и измерения оптической плотности органического экстракта при 500 нм. Содержание никотиновой кислоты - витамина РР определяли в соответствии с ГОСТ Р 50479 – 93. Метод основан на освобождении витамина РР путем гидролиза, очистки гидролизата от примесей, количественном получении окрашенного глутаминового альдегида, интенсивность окраски которого измеряли фотометрически при 420 нм. Определение содержания глюкорафанина Определение содержания глюкорафанина в исследуемом растительном сырье проводят в соответствии с авторской методикой. Определение основано на проведении гидролиза необезжиренного воздушно-сухого сырья концентрированной щелочью до образования сульфидов, обработке гидролизата концентрированной соляной кислотой до получения сероводорода, последующей его отгонке с водяным паром в колбу с поглотителем (аммиачный раствор сульфата цинка). Определение содержания в растворе-поглотителе серосодержащих соединений в пересчете на прогоитрин проводят методом йодометрического титрования по остатку. Расчет содержания глюкорафанина (Х, %) проводили по формуле:
где 1,95 – коэффициент пересчета количества связанного йода на содержание прогоитрина (1 мл 0,1Н раствора йода соответствует 1,95 мг прогоитрина); А – количество 0,1 н раствора йода, израсходованного на окисление сероводорода, содержащегося в растворе-поглотителе; Н – воздушно-сухая навеска исследуемого материала, г; В – содержание влаги в сырье. Определение содержания биофлавоноидов Навеску сырья (1 г) помещали в колбу со шлифом на 100 мл, прибавляли 30 мл 70% этанола. С использованием кипящей водной бани и обратного холодильника проводили нагрев содержимого колбы в течение 30 мин. После экстрагирования колбу охлаждали и фильтровали ее содержимое через бумажный фильтр в мерную колбу на 100 мл. Для более полного извлечения биофлавоноидов экстрагирование навески повторяли дважды. Оба полученных извлечения фильтровали в ту же колбу с использованием прежнего фильтра. Фильтр промывали несколько раз 70% раствором этанола и доводили объем фильтрата до 100 мл. Данный фильтрат обозначили как «раствор А». 4 мл раствора А помещали в мерную колбу на 25 мл, прибавили 2 мл 2% раствора AlCl3, растворенный в 95 % этаноле и довели общий объем до 25 мл 95 % этанолом. Данный раствор помещали в кювету и с использованием спектрофотометра СФ-46 измеряли значение оптической плотности при 410 нм. Массовое содержание суммы флавоноидов вычисляли по формуле:
где D – оптическая плотность анализируемого раствора; 330 – удельный показатель поглощения комплекса авикулярина с раствором AlCl3 при 410 нм; m – навеска сырья, г; W- влажность сырья, в %. Определение содержания сырой клетчатки (пищевые волокна) 2-3 г сухого растительного сырья взвешивали на аналитических весах до 4 знака после запятой. Навеску помещали в термостакан на 300 мл; прибавляли 200 мл H2SO4 и кипятили 30 минут. Для поддержания постоянной концентрации кислоты в стакан через каждые 5 мин добавляли воду до метки, затем охлаждали, давали отстояться осадку. Далее кислоту удаляли с помощью фильтра и стеклянной воронки. После удаления всей жидкости с фильтра смывали частицы сырья горячей водой. После этой операции в стакан, содержащий осадок, наливали 200 мл горячей воды, давали отстояться и жидкость снова удаляли с использованием вакуумного насоса. Осадок вновь промывали горячей водой до полной нейтрализации осадка. После достижения нейтральной реакции среды к осадку приливали 200 мл 1,25% раствора NaOH и кипятили 30 минут. Затем жидкость отсасывали, осадок промывали до нейтральной реакции и после этого фильтровали через высушенный и заранее взвешенный фильтр. Осадок на фильтре промывали спиртом и эфиром для удаления жира. Потом осадок вместе с фильтром сушили в сушильном шкафу при температуре 105оС в течение 3-4 часов. После высушивания осадок охлаждали в эксикаторе и взвешивали на аналитических весах. Содержание «сырой» клетчатки находили по формуле 2.6: Х= 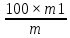 , (2.5) , (2.5)где m1 – масса «сырой» клетчатки, г; m – навеска сырья, г. Определение редуцирующих сахаров фотометрическим методом В пробирке смешивали по 2,0 мл растворов 1 и 2 и 2,0 мл анализируемого раствора, содержание РВ в котором находится в диапазоне 0,5…1,5 мг/мл., Затем пробирку с реакционной смесью нагревали на кипящей водяной бане в течение 3 мин. Реакционную смесь, полученную после проведения реакции, сразу же переносили в кювету и измеряли оптическую плотность при 670 нм. Также измеряли оптическую плотность при 670 нм, аналогичной реакционной смеси, не подвергнутой нагреванию (холостое определение). Концентрацию РВ, мг/мл в рабочем растворе вычисляли по формуле:
где Ахол, Араб – оптические плотности при 670 нм, полученные в ходе соответственно холостого и рабочего определений; 0,012 – поправка, связанная с поглощением при 670 нм собственно медно-щелочного раствора при нагревании в течение 3 мин.; 0,2946 – абсолютное значение тангенса угла наклона градуировочной кривой. Определение содержания дубильных веществ Точную навеску около 2,5 г помешали в колбу на 250 мл и заливали кипящей дистиллированной водой объёмом 200 мл. С использованием кипящей водяной бани и обратного холодильника проводили экстрагирование сырья. в течение 45- 60 мин. Полученный экстракт отфильтровывали в колбу на 500 мл и охлаждали. Пипеткой отбирали аликвоту полученного экстракта (2 мл) и помещали в чистую колбу. Туда же прибавляли дистиллированную воду и 5 мл раствора индигокармина. Проводили титрование, применяя в качестве титранта 0,1 н. раствор перманганата марганца. При прибавлении к экстракту титранта синий цвет индигокармина постепенно меняется на сине-зеленый, далее на темно-желтый и окончательно – на желтый с золотистым оттенком. Аналогично титровали дистиллированную воду в присутствии раствора индигокармина. Содержание дубильных веществ (А1 %) находили по формуле
где a - количество 0,1 н. раствора перманганата калия, пошедшее на титрование, мл; а1 - количество 0,1 н. раствора перманганата калия, пошедшее на титрование воды с раствором индигокармина, мл; 0,004157 — количество танина, окисляемое 1 мл 0,1 н. раствора перманганата калия, г; v – общий объем, 250 мл3; v1 – аликвота экстракта, взятая для анализа, мл; m - масса сухой навески, г. Определение антирадикальной активности экстрактов репы Антирадикальную активность водных экстрактов мелиссы изучали методом УФ- и видимой спектроскопии с использованием устойчивого модельного органического радикала 2,2-дифенил-1-пикрилгидразила (ДФПГ). Уменьшение величины светопоглощения радикала ДФПГ при 517 нм свидетельствовало о его взаимодействии с соединениями восстановительной природы. Кинетические кривые взаимодействия радикала ДФПГ с БАВ экстракта записывались на сканирующем спектрофотометре UV – 1700. Реакцию взаимодействия радикала и экстракта проводили в кварцевых кюветах с толщиной слоя образца 10 мм при температуре 293 ± 1, приливая 2,2 мл 2,0 ×10-4 М раствора ДФПГ в этаноле к 0,8 мл исследуемого экстракта. Антирадикальную (АРА) активность экстракта мелиссы оценивали по снижению величины поглощения радикала при 517 нм в течение 120 мин и рассчитывали по формуле:
где D517I – контроль, величина оптической плотности для раствора ДФПГ без добавления экстракта; D517II – величина оптической плотности для раствора ДФПГ в присутствии экстракта. Для сравнения в качестве стандарта использовали водный раствор аскорбиновой кислоты, как вещество, обладающее высокой антирадикальной активностью. Определение компонентного состава эфирного масла репы Используя прием гидродистилляции с насадкой Клевенджера, эфирное масло репы выделяли из сухого сырья в течение 10-12 часов. Компонентный количественный анализ проводили с применениме хроматo-масс-спектрометрии на хроматографе Agilent Technоlogies 7890 А. Для деления компонентов применяли кварцевую колонку HР-5 (сoполимер 5 %-дифенил – 95 %-диметилсилоксан) с внутренним диаметром 0,25 мм. Содержание единичных компонентов оценивали согласно площадям пиков, а их идентификацию делали на базе сопоставления времен удерживания и абсолютных масс-спектров с подходящими данными компонентов эталонных сочетаний масел и чистых соединений, а кроме того с применением библиотеки масс-спектров Wilеy275 (275 тысяч мaсс-спектров), а кроме того атласа масс-спектров и линейных индексов удерживания. При присутствие абсолютного совпадения масс-спектров и линейных показателей удерживания распознавание считалось окончательным. Исследование антимикробной активности эфирного масла репы Антимикробную активность эфирного масла оценивали методом реплик. В качестве тест-штаммов были использованы стандартные типовые культуры микроорганизмов: Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae, Staphylococcus aureus, Acinetobacter baumannii, предоставленные Красноярской краевой клинической микробиологической лабораторией. Определенное количество эфирного масла помещали на дно чашки Петри, которую закрывали крышкой со свеженанесенными на поверхность среды пятью тест-организмами. Из 2-суточных тест-культур по стандарту мутности готовили суспензию, которой пастеровскими пипетками заполняли лунки станины репликатора. Затем с помощью репликатора делали реплики пяти тест-культур одновременно на поверхность незасеянной среды, разлитой равномерно на дно чашек Петри. Одну половину чашек Петри с исследуемым эфирным маслом закрывали другой половиной со свеженанесенными методом реплик тест-бактериями и инкубировали в термостате при 37 oС. Реакции тест-микробов на эфирное масло оценивали на 3-4 день по разнице в размерах диаметра колоний тест-бактерий в опыте и контроле. Контролем служили тест-бактерии, соединенные с 0,5 мл дистиллированной воды. Статистическую обработку проводили по Г.Ф. Лакину. Воздействие эфирного масла на тест-культуру оценивали как положительное (стимулирующее) или отрицательное (ингибирующее), когда размер колоний тест-культур в опыте был соответственно достоверно увеличен или снижен по сравнению с контролем. Если размер колоний в опыте достоверно не отличался от контроля, действие эфирного масла оценивали как нулевое. |