Глава XIV ОЛИГОФРЕНИИ
(ДИФФЕРЕНЦИРОВАННЫЕ ФОРМЫ)
К дифференцированным формам относят олигофрении с известной этиологией. Число их весьма велико и постоянно увеличивается по мере углубления знаний в области этиологии и клиники олигофрении, а также благодаря использованию современных методов биологических исследований. В связи с этим ниже будут освещены особенности только части наиболее распространенных и достаточно хорошо изученных форм дифференцированных олигофрении.
Исходя из изложенных выше представлений об этиологии олигофрении, мы подразделяем дифференцированные их формы на следующие основные группы: 1) олигофрении при хромосомных болезнях; 2) наследственные формы; 3) смешанные по этиологии (эндогенно-экзогенные) формы; 4) экзо-геиио-обусловлснные формы. Последняя группа разделена на подгруппы в зависимости от преимущественного времени поражения развивающегося мозга: а) связанные с внутриутробными экзогенными поражениями; б) связанные с перинатальной патологией; в) обусловленные ранними постна-тальными экзогенно-органическими поражениями головного мозга.
ОЛИГОФРЕНИИ ПРИ ХРОМОСОМНЫХ БОЛЕЗНЯХ
Хромосомные болезни (синдромы) — клинические состояния, обусловленные нарушением числа или структуры хромосом. Частота хромосомных болезней составляет среди новорожденных 1 : 250 (Е. Ф. Давиденкова, И. С. Либерман, 1975), а по данным R. Turpin, J. Lejeune (1965) — 1 : 100. У эмбрионов частота хромосомных аномалий (аберраций) достигает 4%. С хромосомными аберрациями связано 20% спонтанных абортов (Н. П. Бочков, Н. С. Стонова, 1969). Наиболее характерными клиническими проявлениями аутосомиых аномалий являются признаки психического и физического недоразвития, дисплазии и грубые дефекты телосложения. При заболеваниях, обусловленных аномалиями половых хромосом, слабоумие не является обязательным признаком. Для них более характерно недоразвитие половых желез и нарушение развития вторичных половых признаков. Хромосомные заболевания непрогредисптиы и в большинстве случаев не передаются по наследству.
олигофрении, обусловленные аномалиями аутосом Синдром Дауна (болезнь Дауна)
Заболевание впервые описано J. L. Н. Down в 1866 г. Частота болезни Дауна среди новорожденных составляет 1 : 700 (А. И. Буланов, с соавт., 1967; Н. П. Бочков, 1969) — 1:912 (Е. Ф. Давиденкова, 1965). Она встречается с одинаковой-частотой у детей обоего пола. J. Lejeune, R. Tu'rpin, М. Gau-tier в 1959 г. обнаружили в кариотипе детей с синдромом. Дауна трисомию по 21-й хромосоме. В последующем при патогенетическом исследовании выявлены три варианта аномалий кариотипа: регулярная трисомия, мозанцизм и несбалансированная транслокация.
и
При регулярной трисомии, составляющей около 95% всех
случаев болезни Дауна, в кариотипе выявляется 47 хромосом
(рис. 5). Дополнительная 21-я хромосома обнаруживается во
всех клетках культуры. Кариотип родителей больных с ре-
гулярной трисомией нормальный. Риск повторного рождения
ребенка с болезнью Дауна при трисомии не превышает риска
в популяции и увеличивается с возрастом матери. Около 2%,
случаев болезни Дауна составляют мозаичные варианты, при
которых в организме одновременно обнаруживаются трисом-
ные и нормальные клетки. Механизм возникновения мозаи-
цизма связан с иерасхождепием хромосом в отдельных ядрах^
клеток на ранней ста-
дии деления зиготы,
что приводит к обра- ,
зованию двух клопов
клеток — с 46 и 47 / 2зь-5
хромосомами. Жен-
щины с мозаицизмом с\\ 28 &!£ !?j&>НУ у &
могут иметь как нор- «« ПППй(ш ЙС ft A Ail
мальных детей, так и
больных с регулярной
трисомией по 21-й хро- пкАкААК, V to у х *д мосоме. У некоторых ^»Я«0ЙО вfllДй 6ft
дает в меньшей степе-
ни, чем у больных с
регулярной трисомией.
Риск повторного рож- с т.
ления больного пр6рн Рис* 5* КаРИ0ТИП мальчика 9 лет с синд-
дения оольного реоен- ромом Дауна (трис0мия по 21-й хромо-
ка при мозаичном ва- соме).
больных при мозаи- 13 15 16 1718
цизме интеллект стра-
f 0 ЛД АЛА Jit
192021-22 'XY
рианте такой же, как и при трисомии, а по мнению некоторых авторов (В. И. Гаврилов, 1975), даже меньший.
Транслокационные формы болезни Дауна отмечаются в 3,2% случаев (М. Mickelsen, 1971; S. W. Haug et al., 1967). При этом варианте общее число хромосом в кариотипе 46, так как дополнительная 21-я хромосома транслоцирована на какую-либо другую аутосому. Наиболее часто в транслокации участвуют акроцентрические хромосомы группы D и G, которые чаще соединяются между собой центрическими участками хромосом. Возможны два варианта транслокации — типа D/G и G/G. При транслокационном варианте синдрома Дауна один из фенотипически здоровых родителей может быть носителем сбалансированной транслокации. В кариотипе этих родителей обнаруживается по 45 хромосом, но одна из них состоит из двух; из хромосом группы D и G или из двух хромосом группы G. Поэтому, несмотря на общее число хромосом, равное 45, генотип родителей будет сбалансированным.
Транслокационный вариант болезни Дауна типа D/G в половине случаев имеет наследственный характер и в половине возникает приобретенным путем. Частота рождения детей с синдромом Дауна у матери с транслокацией D/G составляет около 25% всех детей и не зависит от ее возраста. Здесь большее значение имеет возраст бабушки к моменту рождения матери больного ребенка. В данном случае нерасхождение хромосом связывают с нарушением процесса овогенеза, который, как известно, начинается еще во внутриутробном периоде развития женского организма. В пользу этого свидетельствует более старший возраст бабушек к моменту зачатия матерей и тот факт, что носителем хромосом с транслокацией является мать (Е. Ф. Давиденкова, И. С. Либер-ман, 1975; М. S, Newton с соавт., 1972). При транслокации типа G/G наследуется незначительная часть случаев, носителем транслокации G/G чаще является отец (L. Penrose, 1962).
Среди факторов, ведущих к нерасхождению хромосом при болезни Дауна, наибольшую роль играет пожилой возраст матери. Вероятность рождения больного ребенка резко возрастает у женщин старше 35 лет. Механизм нерасхождения хромосом связывают с возрастными изменениями материнского организма (изменение гормонального баланса, нарушение овогенеза, поздний процесс овуляции или поздний процесс оплодотворения), которые влияют на процесс мсйоза (Б. Н. Клоссовский, В. В. Русских, 1956; Л. О. Бадаляп и др., 1971; J. German, 1968). Среди экзогенных факторов, ведущих к перасхождению хромосом, имеют значение ионизирующая радиация, токсические химические вещества, вирусные инфекции.
Клинические проявлении болезни Дауна характеризуются врожденной умственной отсталостью, сочетающейся с рядом типичных аномалий строения, которые делают больных поразительно похожими друг на друга. Заболевание распознается уже при рождении. Дети рождаются с малой массой тела, слабо кричат, вялые, плохо сосут. К наиболее частым нарушениям относятся низкий рост, непропорциональность коротких конечностей и относительно длинного туловища, своеобразное строение черепа и лица. Череп микробрахиоцефаль-ной конфигурации со скошенным затылком. Ушные раковины небольших размеров, деформированные, низкорасположенные. Характерны косой разрез глаз с кожной складкой во внутреннем углу (третье веко, эпикант), наличие участков депигментации на периферии радужки. Нос короткий с широкой уплощенной переносицей (гипертелоризм). Часто отмечаются недоразвитие верхней челюсти, прогнатизм, неправильный рост зубов, высокое («готическое») небо. Язык вследствие гипертрофии сосочков увеличен, складчатый («географический»). К типичным признакам также относятся аномалии строения конечностей: кисть плоская, пальцы широкие, короткие, резко укороченный искривленный кнутри мизинец. Часто выражена непрерывная поперечная складка ладони. На стопах увеличен промежуток между I и II пальцами, иногда отмечается синдактилия.
Частыми признаками болезни Дауна являются деформация костей и нарушение процессов оссификации. Почти у половины больных обнаруживаются пороки внутренних органов, особенно сердечно-сосудистой системы. У всех больных отмечаются нарушения эндокринной системы: недоразвитие половых желез и вторичных половых признаков, снижение основного обмена, ожирение. Следствием этого часто являются сухость и шелушение кожных покровов, ломкость ногтей, волос, алопеция и др. В неврологическом статусе при болезни Дауна обычно выявляются диффузная гипотония, слабость конвергенции, косоглазие, близорукость, нарушения вестибулярного аппарата, признаки вегетативной недостаточности. К характерным внешним проявлениям заболевания относятся своеобразная осанка, опущенные плечи, неуклюжая походка, неловкие движения, низкий глухой голос, маловыразительное лицо, полуоткрытый рот.
В структуре психического недоразвития также отмечается ряд характерных признаков. Умственная отсталость в 75% случаев достигает степени имбецильности, в 20% —идиотии и только в 5% —дебильности (Г. Е. Сухарева, 1965). Мышление больных тугоподвижное, конкретное. Абстрактные понятия, счетные операции, как правило, недоступны. Резко страдает активное внимание, смысловая память. При относительно более сохранной механической памяти больные иногда научаются читать, но.понимание и пересказ содержания для них недоступны. Дети с болезнью Дауна в большей части случаев неспособны к обучению даже по программе вспомогательной школы. У большинства больных отмечается •позднее появление и резкое недоразвитие речи: недостаточное понимание, бедный запас слов, дефекты звукопроизноше-кия. Особенностью психического дефекта являются относительная живость и сохранность эмоциональной сферы. Больные большей частью ласковые, добродушные, послушные. Им не чужды чувства симпатии, смущения, стыда, оби-; ды. Однако некоторые из них бывают раздражительными, упрямыми. Большинство из них любопытны и обладают хорошей подражательной способностью, что способствует привитию навыков самообслуживания и несложных трудовых процессов. Однако, как правило, дети с болезнью Дауна не достигают полной социальной адаптации и нуждаются в постоянной опеке. При мозаицизме чаще встречается более легкая умственная отсталость (В. И. Гаврилов, 1975).
Особенностью возрастной динамики болезни Дауна является позднее половое созревание и раннее появление признаков инволюции (в 30—40 лет). При инволюции больные утрачивают приобретенные навыки, у них нарастает бездеятельность, безразличие (В. В. Русских, 1963; С. Benda, 1960). Многие больные склонны к инфекционным заболеваниям и плохо их переносят. На ЭЭГ выявляют задержку формирования биоэлектрической активности, отсутствие или недостаточность дифференциации ритмов, дизритмию и снижение реактивности. У некоторых больных отмечается пароксизмальная активность, обусловленная дисфункцией базальных структур. При дерматоглифике в большинстве случаев отмечаются характерные изменения рисунка кожного рельефа: непрерывная кожная поперечная складка ладони, наличие одной сгибательной складки на мизинце вместо двух, увеличение числа ульнарных петель, увеличение угла atd>57°.
Патогенез заболевания неясен. Морфологические исследования обнаруживают уменьшение размера и массы мозга, недостаточную дифференциацию борозд и извилин, недоразвитие лобных долей, мозжечка и диэнцефальных отделов мозга, малое количество и неправильное расположение гапг-лиозных клеток коры, нарушение миелинизации. С различным постоянством выявляются изменения в железах внутренней секреции: гипофизе, щитовидной и половых железах, в надпочечниках (В. В. Русских, 1963; С. Benda, 1900). Часто отмечаются комбинированные аномалии развития органов и систем, множественные врожденные уродства. Специфических методов лечения болезни Дауна нет. Показано применение общеукрепляющей и стимулирующей терапии (препараты кальция, железа, алоэ, апилак, поливитамины и др.). Из препаратов стимулирующего действия рекомендуется курсовое лечение витамином Вщ, глютаминовой кислотой, липоцеребрпном, церебролпзииом, а мина лоном, ниами-дом (нуредалом) и долах, соответствующих возрасту. При гормональной недостаточности необходимо тщательное лечение малыми дозами тиреоидина (детям в возрасте моложе* 1 года — 5 мг 1—2 раза в день, на 2-м году — 0,01 -0,03 г ежедневно). Для профилактики болезни Дауна имеет значение медико-генетическое консультирование, способствующее определению риска рождения больного ребенка.
Синдром «кошачьего крика»
Описанный J. Lejeune с соавт. в 1963 г., имеет наиболее очерченные клинические проявления среди других синдромов, обусловленных структурными аномалиями аутосом. Синдром чаще наблюдается у лиц женского пола, его частота в популяции не уточнена. Типичные клинические проявления достаточно стабильны. Отмечаются задержка физического развития и врожденные диспластическис признаки: круглое лицо, «антимонголоидный» разрез глаз, гииертелоризм, эиикант, низкое расположение и асимметрия ушных раковин. Реже встречаются микроцефалия и пороки развития внутренних, органов. Патогномоничным проявлением, которое способствовало выделению синдрома, являются аномалия развития-гортани, вследствие чего дети имеют особый, напоминающий: кошачий («мяукающий») тембр голоса. При дерматоглифи-ческом исследовании па ладони обнаруживается непрерывная поперечная складка.
Степень психического недоразвития варьирует в широких пределах, но чаще имеет место имбецильность и идиотия. Диагноз ставится на основе типичных клинических проявлений и данных цитогенетического исследования (обнаружение делеции короткого плеча 5-й хромосомы в группе В). Кроме того, у части больных обнаружены транслокации типа 5/D и 5/G, которые могут иметь наследственное происхождение (J. Philip et al., 1969). В случае обнаружения транслокации показано цитогенетическое обследование родителей.
ОЛИГОФРЕНИИ ПРИ АНОМАЛИЯХ ПОЛОВЫХ ХРОМОСОМ
Синдром Шерешевского—Тернера
Синдром описан Н. А. Шерешевским в 1925 г. и Н. Turner в 1938 г. Распространенность в женской популяции составляет 0,3 на 1000. Его частота резко возрастает среди низкорослых женщин с недоразвитием вторичных половых признаков и первичной аменореей. Клинические проявления синдрома отмечаются уже с рождения. У новорожденных девочек обнаруживается малые масса тела и рост, лимфатический отек на кистях и стопах вследствие аномалий развития лимфатических сосудов. На коже нередко отмечаются витили-го, пигментированные пятна, гемангиомы и нейрофибромы. Шея короткая с избыточной кожей на заднебоковой поверхности, которая примерно у половины больных выступает в виде шейной складки. Нередко обнаруживаются аномалии развития внутренних органов, чаще — пороки сердца, коарк-тация аорты, стеноз легочной артерии, аномалии почек и др.
Врожденные аномалии строения придают больным своеобразный вид: «антимонголоидный» разрез глаз (наружные углы глаз расположены ниже внутренних), эпикант, низкое расположение ушей, короткая и широкая шея с низким уровнем роста волос придают им старческий вид. Отмечается также нарушение строения скелета: деформация грудной клетки, широкая ладонь, клинодактилия мизинцев, укорочение пальцев с поперечной исчерченностью ногтей, вальгусное положение коленных суставов, деформация стоп, реже синдактилия и полидактилия. Нередко обнаруживается сращение и укорочение позвонков и spina bifida. С возрастом появляется значительное отставание в росте, который, как правило, не превышает 150 ем. Нарастают диспропорции телосложения: преобладание верхней части туловища, широкие плечи, узкий таз, укорочение нижних конечностей. Конституция строения девочек приближается к мужской. В препубертатном и пубертатном возрасте выявляются признаки полового инфантилизма. Наружные половые органы недоразвиты, иногда отмечается гипертрофия клитора. Молочные железы не развиты, соски втянуты, широко расставлены. Оволосение лобка и подмышечных впадин отсутствует или скудное (Д. К. Берлинская, 1965).
Патогномоничными признаками являются аномалии строения внутренних половых органов и гонадальный дисге-нез: узкое длинное влагалище, недоразвитая матка, отсутствие или фиброзное перерождение яичников. В большинстве случаев отсутствуют примордиальные фолликулы. Один из важных признаков заболевания — первичная аменорея. Однако у некоторых больных могут отмечаться редкие и скудные менструации. Интерес к противоположному полу значительно снижен. Умственное недоразвитие обнаруживается у незначительной части больных, чаще выражено нерезко, но изредка достигает степени идиотии. У многих больных е. возрастом появляется критика к своему состоянию. Обычно трудолюбивые и благодушные, они становятся более замкнутыми, раздражительными, появляется склонность к невротическим реакциям.
На ЭЭГ нередко отмечаются признаки задержки коркового электрогенеза. дизритмия. При рентгенологическом исследовании выявляется задержка окостенения, нарушение слияния эпифизов с метафизами, остеопороз трубчатых костей. В пубертатном периоде при биохимическом исследовании обнаруживается повышенное содержание гонадотропи-нов и снижение уровня эстрогенов.
Дерматоглифическое исследование выявляет более близкое расположение к центру аксиального трирадиуса и увеличение угла atd.
* Половой хроматин определяется в клетках эпителия слизистой
рта.
При цитогенетическом исследовании в типичных случаях в хромосомном наборе больных выявляется 45 хромосом (45/ХО) — 22 пары аутосом и только одна Х-хромосома. В отдельных случаях при синдроме Шерешев-ского — Тернера может обнаруживаться мужской кариотип (46/XY). Проявление этой аберрации женским фенотипом объясняется полным отсутствием функциональной активности Y-хромосомы. В то же время выраженные типичные признаки синдрома Шерешевского — Тернера могут встречаться и у мужчин с нормальным кариотипом (46/XY) (Е. Ф. Дави-денкова, И. С. Либерман, 1975). Несоответствие кариотипа и фенотипа предположительно объясняется тем, что вначале имелась хромосомная мозаика (45, ХО/46, XY), но после того, как половая дифференциация пошла по мужскому типу, клон ХО элиминировался. В кариотипе больных могут выявляться более сложные формы мозаицизма (45, ХО/46, XY; 45, XO/47/XYY). Эти варианты хромосомных аберраций могут проявляться как женским, так и мужским фенотипом или гермафродитизмом. Характерной отличительной особенностью этих вариантов синдрома Шерешевского — Тернера является отсутствие полового хроматина в ядрах клеток. В настоящее время, в соответствии с теорией М. Lyon (1961), известно, что в женском организме (кариотип 46, XX) функционирует только одна Х-хромосома, а другая находится в инактивном состоянии и дает тельце полового хроматина (тельце Барра). Поэтому в норме половой хроматин содержится примерно в 80% ядер женских соматических клеток и отсутствует или не превышает 5% в мужских клетках, кариотип которых имеет одну Х-хромосому (46, XY). Сколько бы ни прибавлялось в хромосомном наборе Х-хромосом, активной остается одна, а все лишние представлены в виде полового хроматина. При структурных аномалиях Х-хромосо-мы происходит изменение телец Барра. Отсюда ясно, что при хромосомных аберрациях количество и размеры телец полового хроматина находятся в зависимости от количества и структуры Х-хромосом*. В зависимости от отсутствия или наличия лишних телец Барра различают хроматппотрица-тельиые и хроматинположительные хромосомные аномалии. К хроматинположительным вариантам синдрома Шерешевского— Тернера, составляющим около 20%, относятся случаи с нормальным кариотипом (XX), когда одна Х-хромосо-ма морфологически изменена, мозаичные варианты — ХО/ХХ, ХО/ХХ/ХХХ и другие более сложные разновидности. При мозаицизмс ХО/ХХ половой хроматин обнаруживается в меньшем проценте случаев, чем в норме, а при ХО/ХХ/ХХХ в некоторых клетках находят по два тельца Барра. Если лишняя хромосома, что бывает нередко, структурно изменена (траиелокация, делеция, изохромосома и др.), половой хроматин также изменен в размере (Е. Ф. Давиденкова, И. С. Либерман, 1975).
11ри различных вариантах хромосомных аномалий синдрома Шерешевского — Тернера клинические проявления широко варьируют. Можно считать закономерностью, что с увеличением количества лишних хромосом степень психического недоразвития утяжеляется. В литературе имеются единичные наблюдения семейных случаев синдрома Шерешевского — Тернера. Диагноз ставится на основании клиники и лабораторных данных (изменения полового хроматина и аномалии половых хромосом). Лечение при синдроме Шерешевского — Тернера состоит п применении гормональных препаратов (эстрогенов) в преиубертатном возрасте.
Синдром «трипло-Х»
Синдром «трипло-Х» впервые описан P. A. Jacobs с соавт. в 1959 г. Это пограничное между нормой и патологией состояние характеризуется наличием в кариотипе более двух Х-хромосом (обычно трисомия X) и увеличением количества телец полового хроматина. Согласно данным Е. Ф. Давиден-ковой и И. С. Либерман (1975), частота трисомии X составляет среди новорожденных девочек и женщин 1 : 1000 (0,1%), а среди умственно отсталых 0,59%. Большинство девочек и женщин с синдромом трисомии X выявлено среди больных психиатрических больниц. Клинические проявления синдрома полиморфны. У части пациентов с трисомией X не обнаруживается каких-либо признаков нарушения половой дифференциации, отклонений в физическом и психическом развитии, они могут иметь здоровых детей с нормальным кариотипом. Одним из проявлений трисомии X является неглубокая умственная отсталость, которая отмечается у 75% больных (Е. Ф. Давиденкова, И. С. Либерман, 1975). Особое внимание привлекает тот факт, что при наличии добавочной Х-хромосомы резко возрастает частота заболевания шизофренией (Ю. 11. Филиппов, 1971; Т. Raphael, М. W. Shaw, 1963).
У многих больных с трисомией X наблюдаются задержка физического развития, негрубые диспластические признаки: эпикант, высокое твердое небо, клинодактилия мизинцев. Реже встречаются больные высокого роста. У некоторых больных отмечается бесплодие в связи с недоразвитием фолликулов и расстройством их нормальной функции, что ведет к эндокринному дисбалансу, аменорее и преждевременному климаксу. Цитогенетические исследования при трисомии X выявляют 47 хромосом (47, XXX) и двойной половой хроматин. Значительно реже обнаруживается более сложная по-лисомия X: тетрасомия (ХХХХ) и пентасомия (ХХХХХ) с соответствующим увеличением количества телец полового хроматина. В этих случаях степень психического недоразвития выражена резче и коррелирует с количеством дополнительных Х-хромосом. В качестве симптоматического лечения при синдроме трипло-Х применяются гормональные препараты.
Синдром Клайнфелтера
Синдром Клайнфелтера (47, XXY) описан Н. F. Klinefelter, Е. С. Reifenstein, F. J. Albright в 1942 г. Его частота в мужской популяции составляет в среднем 0,2%, среди умственно отсталых— 1—2%,асреди мертворожденных — 3,4% (Н. П. Бочков, 1966; Л. О. Бадалян и др., 1971; Е. Ф. Давиденкова, И. С. Либерман, 1975, и др.). Клинические проявления синдрома крайне вариабельны: от внешне нормального физического и интеллектуального развития до выраженного евнухоидизма и глубокой дебильности. В ряде случаев уже в раннем детском возрасте отмечаются отдельные симптомы со стороны физического развития: узкий и низкий лоб, густые и жесткие волосы, узкая, плоская грудная клетка, высокое стояние таза, недоразвитие половых органов, евнухоидные пропорции (А. М. Пономаренко, 1965). Более типичные симптомы заболевания отчетливо начинают обнаруживаться в пубертатном возрасте. Для фенотипа больных характерны высокий рост, астеническое сложение, узкие плечи, удлиненные конечности, слаборазвитая мускулатура. Примерно у половины больных отмечается гинекомастия и евнухоидные признаки: скудная растительность на лице и в подмышечных впадинах, широкий таз, ожирение и оволосение на лобке по женскому типу. Выделяются два типа телосложения: для одних больных характерен высокий рост и астенические черты телосложения, для других — евнухоидные пропорции и гинекомастия, которая может быть одно- или двусторонней. В более редких случаях отмечаются аномалии строения зубов, скелета и конечностей. Часто встречается ожирение по женскому типу.
Патогномоничными признаками синдрома Клайнфелтера являются недоразвитие половых органов и бесплодие. Гонады больных уменьшены в размерах (микроорхидизм), отмечается атрофия и гиалинизация семявыводящих канальцев, дегенерация лейдиговских клеток и избыток фиброзной ткани. В пубертатном периоде в связи с отсутствием сперматогенеза и недостаточностью функции интерстициальных клеток обнаруживается снижение секреции 17-КС и повышение продукции гоиадотропинов передней доли гипофиза. В неврологическом статусе в ряде случаев имеются мышечная гипотония и диэнцефально-вегетативные расстройства, в т. ч. приступообразные; часто встречается .моторная недостаточность.
Умственное недоразвитие при синдроме Клайнфелтера чаще выражено нерезко, но в отдельных случаях достигает степени глубокой дебильности и выявляется уже в раннем детском возрасте. В то же время встречаются случаи заболевания с практически нормальным интеллектом. В качестве особенностей структуры интеллектуального дефекта в детском возрасте у большинства больных можно отметить сочетание интеллектуальной недостаточности с относительно более глубокой незрелостью эмоционально-волевой сферы, которая по своим проявлениям приближается к психическому инфантилизму. У этих больных, наряду с недостаточностью внимания, восприятия, памяти и абстрактного мышления, более резко и рельефно обнаруживаются чрезмерная внушаемость, подражательность, подчиняемость, недостаточность самостоятельности, чрезмерная привязанность к близким, нередко с элементом назойливости. Настроение обычно повышенное, с эйфорическим оттенком, имеет тенденцию к беспричинным колебаниям, иногда отмечается склонность к эксплозивным аффективным вспышкам. У таких больных рано проявляется недостаточное чувство долга, ответственности, активности и неспособность к длительному волевому усилению и напряженной деятельности. Эти особенности эмоционально-волевой сферы как бы выступают на первый план в структуре олигофренического дефекта и утяжеляют общую клиническую картину психического недоразвития при синдроме Клайнфелтера.
У части больных, преимущественно при наличии легкого психического недоразвития, с началом обучения в школе и особенно в пубертатном и постпубертатном возрасте появляется сознание своей неполноценности, которое становится источником внутреннего конфликта. У больных начинает преобладать гипотимический фон настроения, нередко с раздражительностью, легко возникают невротические и патохарактерологические реакции. В литературе также описываются случаи синдрома Клайнфелтера с явлениями апатии, адинамии, депрессивными, ипохондрическими, навязчивыми, нарко-

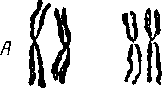
4-5
Ш IX и п и п и
в-12
в шш £ и п Ы
13-15№ Г/ 18
13-го
f ХШ G **** $)К
ft
21-22X X Y
Рис. 6. КариотИ 'П мальчика 13 лет с синдромом Клайнфелтера. лептическими и шизофреноподобиыми расстройствами (М. М. Райская, 1972; Н. Forssman, G. Hambert, 1963, и др.). При ЭЭГ выявляются задержка формирования основных корковых ритмов, преобладание медленных высокоамплитудных колебаний в передних отделах коры. Изменения биоэлектрической активности коррелируют со степенью интеллектуального недоразвития. Дерматоглифические изменения при синдроме Клайнфелтера характеризуются изменением пальцевых узоров в виде увеличения ульнарных петель и более проксимального расположения трирадиуса. Цитогенетические исследования обнаруживают в ядрах клеток содержание полового хроматина, соответствующее женскому типу. Карио-логическое исследование выявляет 47 хромосом (47, XXY) (рис. 6.). Реже встречаются варианты синдрома с кариоти-пом 48, XXXY и 49, XXXXY, соответственно с двойным и тройным половым хроматином, а также варианты с дополнительной Y-хромосомой (48, XXYY), различные формы мозаи-цизма и другие хромосомные аберрации. Как правило, степень интеллектуального недоразвития выражена тем глубже, чем больше дополнительных половых хромосом в кариотипе. Специфического лечения заболевания не существует. В качестве симптоматической терапии применяются гормональные препараты: прогестерон, эстрадиол-пропионат, тестосте-рон-пропионат и др. Однако гормональная терапия в отношении бесплодия и гинекомастии малоэффективна. Анд-рогены в некоторой степени влияют на улучшение психического развития. Лечение гинекомастии в основном проводится хирургическим путем. В комплексе лечебных мероприятий определенная роль отводится рациональной психотерапии для устранения вторичных невротических и патохарактерологических реакций. Синдром XYY Синдром описан у мужчин высокого роста с антисоциальным поведением. Многие из них отстают в психическом развитии (P. Jacobs et al., 1965, 1968). Частота синдрома составляет среди новорожденных около 1 : 1000 (Е. Ф. Давиденкова, И. С. Либерман, 1975). Значительно более высокие цифры (1 : 250) приводят американские авторы (F. Segrovich et al., 1969). При рождении детей с синдромом 47, XYY каких-либо отклонений от нормы не выявляется, но нередко уже в дошкольном возрасте отмечается превышение нормы массы тела и роста, нарушение формирования речевых функций, задержка психического развития и признаки моторной недостаточности с явлениями двигательной возбудимости (Л. А. Петров и др., 1976). Клинические проявления синдрома полиморфны и малоспецифичны. Наиболее частым признаком является высокий рост, который у взрослых составляет в среднем 186 см (рис. 7, а). Однако этот признак не является абсолютным, так как имеются описания мужчин среднего роста с карио-типом 47, XYY. У части больных отмечаются нерезко выраженные евнухоидные черты телосложения и диспластические признаки: неправильное строение зубов, увеличение нижней челюсти, аномальный прикус, девиация коленных и локтевых суставов, радиоульнарный синостоз, spina bifida. Иногда обнаруживается повышение уровня андрогенов и лютеинизиру-ющего гормона. Половая функция у этих больных не нарушена и они имеют здоровых детей с нормальным кариотипом (Е. Ф. Давиденкова, И. С. Либерман, 1975). Примерно 80% больных с синдромом XYY имеют легкие признаки психического недоразвития с неравномерной своеобразной структурой интеллектуального дефекта. У этих больных в большей степени страдают предпосылки интеллектуальной деятельности, рано обнаруживаются дисгармония эмоционально-волевой сферы и формирование аномальных качеств личности при негрубом недоразвитии абстрактного мышления. В раннем возрасте эти дети мало пользуются речью и обнаруживают признаки аутистического поведения. Они малообщительны, замкнуты, плохо сходятся с детьми, не проявляют глубоких привязандостей к близким. В школьном возрасте более отчетливо проявляются неустойчивость внимания, неусидчивость, неспособность к длительному интеллектуальному напряжению и целенаправленной трудовой деятельности. Эмоционально-волевые нарушения выражаются в бес
v и
i) (г
4 5
7 г- *
щ о и w м к
6-12
питГоHi1920е ПЬА161718
ку г
21 iV д
^^^^^^^^^^^^^^
Рис. 7. Синдром XYY у мальчика 11 лет (а) и его кариотип (б).
причинных колебаниях настроения, взрывчатости, импульсивности и агрессивности по незначительному поводу. В то же время больные внушаемы, легко имитируют поведение окружающих. Дети и подростки с синдромом XYY при конфликтных ситуациях часто дают эксплозивные реакции с агрессией, совершают побеги из школы и дома. У некоторых больных отмечается наклонность к воровству, поджогам и другим правонарушениям. Эти дети и подростки могут легко усваивать программу вспомогательной школы, но их школьная и трудовая адаптация нарушена в связи с выраженной патологией поведения. При цитогенетическом обследовании с помощью люминесцентной микроскопии в буккальных мазках обнаруживается Y-хроматин. При анализе кариотипа выявляется дополнительная Y-хромосома (см. рис. 7,6). Специфического лечения синдрома не существует. Показано применение седативных средств. Основное же значение имеет коррекционно-воспитательная работа, а в более старшем возрасте — рациональная психотерапия. |
 Скачать 1.63 Mb.
Скачать 1.63 Mb.