Системы крови
 Скачать 7.82 Mb. Скачать 7.82 Mb.
|

Классификация анемийОна основана на патогенетических признаках с учетом важнейших этиологических факторов, морфологических изменений эритроцитов, способностей кроветворной к ткани к регенерации и восстановлению количества и качества эритроцитов и Hb в крови и т.д. Различают следующие основные виды анемий:
Постгеморрагические анемииОстрая постгеморрагическая анемия (ОПГА) развивается в результате быстрой и массивной, обычно разовой потери крови (за счет наружного или внутреннего кровотечения). Этиология. Причинами развития ОПГА являются: травматическое или язвенное повреждение различных кровеносных (главным образом, крупных артериальных) сосудов тела (шеи, желудка, кишок, матки и др.). Клиника. В развитии ОПГА выделяют 3 стадии: 1) рефлекторная, 2) гидремическая, 3) регенераторная. Первая, рефлекторная, стадия развивается сразу после кровотечения. Ее основу составляет спазм сосудов, приводящий к понижению объема сосудистого русла на фоне гиповолемии простой, сопровождающейся равномерным уменьшением плазмы, Нb и эритроцитов (проявляется нормальными величинами цветового показателя и количеств эритроцитов и Нb в единице объема крови, то есть развивается нормохромная нормоцитарная анемия). Вторая, гидремическая, стадия развивается через 2-3 дня после остановки кровотечения. В этот период ослабевает дефицит ОЦК вследствие мобилизации из внесосудистого пространства воды и белков, что сопровождается разжижением крови (количество эритроцитов и Нb в единице объема крови понижается, т.е. возникает относительная эритропения). Одновременно отмечается усиление разрушения части эритроцитов за счет активизации фагоцитирующих мононуклеаров, приводящее к развитию абсолютной эритропении. Третья, регенераторная, стадия развивается после 4-5 дней после прекращения кровопотери. Характеризуется активизацией эритропоэза, обусловленной, главным образом, повышением продуктов распада эритроцитов и увеличением образования эритропоэтина. В эту стадию отмечается повышение содержания в крови ретикулоцитов и появление в ней нормобластов. При этом регенерация эритроцитов опережает их созревание. Удельное содержание Нb в каждом эритроците понижается (что проявляется снижением цветового показателя крови). Полное восстановление основных параметров системы красной крови после острой кровопотери происходит через 30 и более суток Хроническая постгеморрагическая анемия (ХПГА) развивается, главным образом, в результате длительных, часто повторяющихся небольших кровопотерь из-за повреждения различных по размеру и структуре кровеносных сосудов, особенно, органов внешнего дыхания, желудка, пищевода, почек, матки и др., а также нарушений тромбоцитарно-сосудистого и коагуляционного видов гемостаза (например, при геморрагическом диатезе). Данный вид анемии характеризуется снижением (менее 0,86) цветового показателя крови, развитием микроцитоза и т.д. При ХПГА обычно после активизации наблюдается прогрессирующее снижение регенераторной способности красного ростка костного мозга. Развивается гипохромная микроцитарная анемия. Картина периферической крови представлена на схеме 23-5. Гемолитические анемииВ основе гемолитической анемии (ГА) лежит гемолиз эритроцитов различного происхождения (наследственный и/или приобретенный, внутриклеточный и/или внутрисосудистый). ГА проявляется уменьшением числа эритроцитов и продолжительности их жизни, снижением дыхательной функции крови, развитием гемоглобинемии, повышением цветового показателя, а также развитием гемолитической желтухи, избыточной продукции желчных пигментов (проявляющейся развитием гипербилирубинемией, особенно за счет увеличения в плазме свободного, непрямого билирубина; повышением содержания стеркобилина в кале и стеркобилиногена и уробилиногена в моче); уменьшением уровня гаптоглобина в плазме, а значит и связывания Hb, сопровождающегося увеличением количества Hb в плазме (при величинах свыше 25%), появлением и повышением уровня Hb в моче, развитием гемосидероза (отложения в тканях гемосидерина- темно-желтого железосодержащего пигмента, усиленно образующегося с участием макрофагов при распаде гемоглобина эритроцитов) и спленомегалии. Особенности гематологических проявлений при ГА: - резкое увеличение в костном мозге и крови содержания ретикулоцитов (до 5-10-20% и выше ) и ядросодержащих эритроцитов (нормобластов, пронормобластов и даже эритробластов ), обусловленных продукцией эритро- поэтина; - наличие преимущественно определенной формы эритроцитов при том или ином виде анемии. Так, при серповидноклеточной анемии определяются серповидные эритроциты (дрепаноциты), при наследственном овалоцитозе - овалоциты, при наследственном микросфероцитозе (болезни Минковского-Шоффара) - микросфероциты, при талассемии - мишеневидные эритроциты, при действии гемолитических ядов или наследственной энзимопатии - патологические включения в эритроцитах в виде телец Гейнца, при различных, особенно экзоэритроцитарных гемолитических анемиях выявляются выраженный анизоцитоз и т.д.; - развитие нормохромной или гиперхромной анемии; - уменьшение времени кругооборота плазменного железа; - развитие нейтрофильного лейкоцитоза и тромбоцитоза, вызванных гипертрофией гранулоцитарного и метакариоцитарного ростков костного мозга, обусловленного действием соответствующих макрофагальных КСФ, усиленно образующихся при интенсивном гемолизе эритроцитов. Приобретенные ГА (экзоэритроцитарные) Возникают в результате действия на эритроциты повреждающих веществ (ядов), находящихся в плазме. К ним относятся: токсины (растительного и животного происхождения); микроорганизмы (гемолитический стрептококк, анаэробные бактерии и др.); паразиты (малярийные плазмодии, лейшмании и др.); химические вещества (соли тяжелых металлов, соединения мышьяка, тринитротолуол и др.); лекарства (фенацетин, хинин, сульфаниламиды, пенициллин, стрептомицин, ПАСК, фтивазид и др.); пищевые продукты (конские бобы, стручковые растения). Может возникать механический гемолиз (при функционировании искусственного клапана сердца, длительном марше, беге). Гемолиз эритроцитов происходит также при диабете, уремии, гипофосфатемии (отмечаемой особенно при алкоголизме), дефиците 2,3-дифосфоглицерофосфата и АТФ или при избытке кислых продуктов (лактацидемии). Кроме того, выделяют иммунную ГА. Последняя может быть двух основных видов: изоиммунной (возникает при переливании видо-, группо-, резуснесовместимой крови, а также при наличии у матери и плода различных групп крови в системе АВО как при наличии, так и отсутствии Rh-фактора в крови) и аутоиммунной (образование антител против собственных эритроцитов в результате срыва иммунологической толерантности к собственному белку). В основе патогенеза гемолиза лежат: 1) метаболические или структурные повреждения мембран эритроцитов (активизация процессов ПОЛ и др.); 2) увеличение осмоляльности внутриклеточного содержимого эритроцитов; 3) снижение способности эритроцитов к деформации (обычно происходящей в микрососудах органов). Картина периферической крови представлена на схеме 23-5. Наследственные ГА (эндоэритроцитарные) Обусловлены генетическими нарушениями морфологии эритроцитов (мембранопатии), метаболизма этих клеток (энзимопатии) и синтеза гемоглобина (гемоглобинопатии). Наследственные мембранопатии. Основным патогенетическим звеном ГА в результате мембранопатий являются наследственные изменения в первичной последовательности аминокислот одного из структурных мембранных белков эритроцитов (в частности, отсутствия белка спектрина) и дефекты липидных компонентов мембран этих клеток. Среди них наиболее часто встречается наследственный микросфероцитоз (болезнь Минковского-Шоффара), который наследуется по аутосомно-доминантному типу. Заболевание чаще развивается в юношеском, молодом и зрелом возрасте, хотя может возникать и у детей. Патогенез данного вида анемии представлен на схеме 23-1. 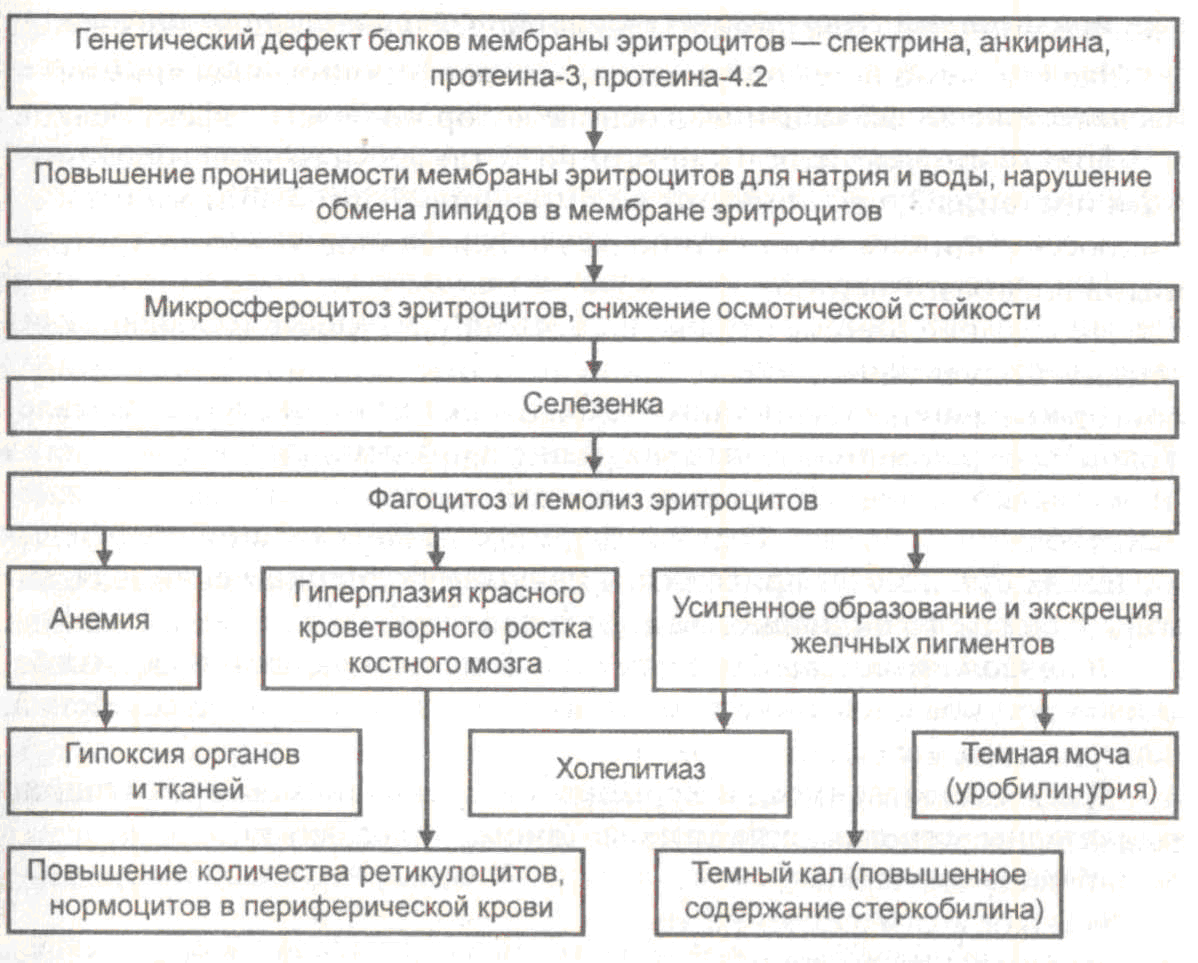 Схема 23-1. Патогенез наследственной микросфероцитарной анемии (Окороков А.Н., 2001) Эритроциты обычно становятся гиперхромными. Генетический дефект мембраны эритроцита (потеря мембранных липидов и др.) приводит к повышению ее проницаемости для ионов натрия и избыточному поступлению в клетку воды. Эритроциты набухают, изменяют свою форму, становятся сфероцитами с диаметром около 6 мкм и теряют эластичность. В результате нарушается способность эритроцитов деформироваться в мелких кровеносных сосудах (то есть, уменьшается пластичность эритроцитов). При этом отмечается отрыв частей оболочек микросфероцитов с развитием клеточной фрагментации и последующей их деструкцией, особенно, при прохождении их через эти микрососуды. Резко выраженное укорочение продолжительности жизни микросфероцитов (до 12-14 дней) связано также со снижением энергозапасов в них, так как значительная часть энергии расходуется для удаления избытка воды и натрия из этих клеток. В развитии сфероцитоза важное значение придают селезенке, так как спленэктомия резко уменьшает сфероцитоз. Наследственные мембранопатии могут также проявляться развитием овалоцитоза, стоматоцитоза, акантоцитоза и т.д. Наследственные энзимопатии. Обусловлены наследственным дефицитом, главным образом, ферментов пентозофосфатного цикла, аэробного гликолиза и системы глутатиона. Чаще выявляется энзимопатия, связанная с наследственным дефицитом глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ), которая, как известно, катализирует начальный этап пентозофосфатного шунта. Заболевание развивается по рецессивному типу, сцепленному с Х-хромосомой, у каждого 10 мужчины, особенно в субтропиках и тропиках. При дефиците Г-6-ФДГ отмечается блокада окисления глюкозо-6-фосфата, главным образом, в пентозофосфатном цикле. Страдает также аэробный гликолиз. Это приводит к нарушению процессов восстановления НАДФ в НАДФН2 и снижению образования восстановленного глутатиона в эритроцитах. Последний защищает SH-группы глобина и мембраны эритроцитов от оксидантного повреждения, в частности, от окислительного действия перекисей и свободных радикалов, образующихся при различной инфекционно-токсической патологии, в том числе при приеме лекарств-окислителей (противомалярийных средств: хинина, делагила; сульфаниламидов, нитрофуранов; производных салициловой кислоты: аспирина; анальгетиков: анальгина, амидопирина; противотуберкулезных средств: ПАСК, фтивазида; и др.). При употреблении в пищу конских бобов и стручковых растений, а также при гриппе и вирусном гепатите может развиваться гемолитический криз. Активизация процессов перекисного окисления гемоглобина и липидов мембран эритроцитов повышает проницаемость последних. Это сопровождается нарушением содержания ионов в эритроцитах, снижением их осмотической резистентности, развитием разрушения гемоглобина и в конечном итоге - острого внутрисосудистого гемолиза. Характерным признаком данного вида анемии является образование в эритроцитах телец Гейнца (денатурированных фрагментов осевшего гемоглобина), которые закрепляясь на внутренней поверхности оболочки в еще большей степени снижают ее эластические свойства. Образование телец Гейнца сопровождается повышением концентрации метгемоглобина в неполноценных эритроцитах. Выявляют также энзимопатии, связанные с наследственным дефицитом ферментов гликолиза (пируваткиназы, гексокиназы, фосфофруктокиназы и др.) и негликолитических энзимов (глутатион пероксидазы, глутатион синтетазы, глутатион редуктазы).Недостаток данных ферментов нарушает мобилизацию глюкозы и выработку энергии в эритроцитах, что приводит к нарушению ионного состава в этих клетках и снижению их осмотической резистентности и продолжительности жизни. Наследственные гемоглобинопатии (гемоглобинозы). Это заболевания, в основе которых лежит нарушение синтеза гемоглобина, возникающее в результате наследственно обусловленной аномалии образования глобина, особенно в странах с жарким климатом. Как известно, у здорового человека HbA1 (около 97 %) содержит две - и две -цепи, HbA2 (около 2 %) - две - и две - цепи, HbF (около 1 %) - две - и две -цепи. Выделяют качественные и количественные Hb-патии. Качественные гемоглобинопатии. К ним относят заболевания, связанные с генетическим дефектом структуры (например, аминокислотной последовательности) той или иной цепи глобина (, , , ). Среди них наибольшее клиническое значение представляет гемоглобиноз S или серповидно-клеточная анемия. Данная анемия наследуется с неполным доминированием вследствие мутации структурного гена и синтеза в результате этого не HbA, а HbS, в -цепи которого у места связывания гена и глобина, гидрофильная глутаминовая кислота заменена гидрофобным валином. В процессе перехода от окисленной к восстановленной форме изменяется заряд патологического HbS, уменьшается его растворимость, происходит образование кристаллических агрегатов, что и лежит в основе серповидности эритроцитов. Серповидные эритроциты (или дрепаноциты) повышают вязкость крови, замедляют кровоток, вызывают стаз, что в еще большей степени углубляет гипоксию. Эти клетки имеют короткий период жизни и быстро разрушаются. Изменение формы эритроцитов и снижение эластичности их мембран определяет повышенное разрушение этих клеток в организме (приемущественно в селезенке, печени) У гетерозигот по HbS (встречается у 8-13% негров) развивается средней или легкой степени тяжести анемия. Это обусловлено тем, что у них эритроциты содержат 20-45% HbS и 55-80% HbА1. У гетерозигот серповидные эритроциты подвергаются гемолизу, как правило, в ответ на развитие различных видов гипоксии и действие на них различных повреждающих факторов. При гетерозиготной по HbS анемии больные живут относительно долго. В отличие от этого, у гомозигот по HbS (встречается у 0,3% людей) эритроциты имеют только HbS. У таких пациентов серповидные эритроциты в крови разрушаются даже при нормальном рО2 . У них развивается тяжелая гемолитическая анемия, вплоть до развития гемолитических кризов. Такие больные погибают быстрее, они редко доживают до 40 лет. Количественные гемоглобинопатии. К ним относят наследственные заболевания, в основе которых лежит частичное или полное блокирование, или увеличение синтеза одной из полипептидных (чаще - или -) цепей глобина. При блокадах этих цепей наиболее часто развивается талассемия (средиземноморская анемия). Блокада синтеза -цепей глобина проявляется в виде развития -талассемии. При данной анемии снижается синтез в костном мозге и содержание в крови основного физиологического HbA1, в то время как уровень HbF и HbA2 напротив возрастает. Отмечается уменьшение срока жизни эритроцитов, развиваются хроническая гипохромная анемия и спленомегалия, появляются мишеневидные эритроциты, количество железа в организме обычно в норме или больше нормы. Данная анемия воникает в результате одинаковой или разной степени выраженности мутации одного или двух генов, находящихся в соотвествующих локусах β-глобина на хромосоме 11. В зависимости от степени дефицита β-цепей глобина клинически различают большую β-талассемию (болезнь Кули), промежуточную и малую. Развернутая картина -талассемии (большая талассемия) возникает при гомозиготной мутации двух генов, что приводит к наибольшему дефициту синтеза β-цепей. При промежуточной β-талассемии выявляется одна слабая, а другая – тяжелая мутация двух генов в соответствующих локусах на 11 хромосоме. При слабой β-талассемии отмечается мутация только одного гена в этой же хромосоме. Снижение синтезa-цепей глобина в эритроцитах, возникающее при наследственной патологии соответствующих регуляторных генов обуславливает развитие -талассемии. Последняя чаще (в 80-85% случаев) возникает в результате мутации одного или нескольких из четырех генов, находящихся в соотвествующих локусах α-глобина на хромосоме 16. клинические проявления коррелируют со степенью нарушения синтеза α-глобиновых цепей. При наибольшей тяжести заболевании угнетается синтез всех нормальных видов гемоглобина, имеющих -цепь (то есть и HbA1, и HbA2, и HbF). Избыточное образование -цепей глобина вызывает у людей образование патологического HbH, который обладает высокой неустойчивостью, легко оседает в клетках, образуя “тельца включения”, и почти полностью не выполняет кислородтранспортной функции. При этом развивается активный гемолиз эритроцитов в макрофагах печени и селезенки. Наследуется данный вид гемоглобинопатии по аутосомно-доминантному типу. Избыточное образование -цепей глобина способствует появлению нестабильного гемоглобина, который преципитирует и выпадает в центре эритроцита в виде “телец включения”, придавая им форму мишеней. Кроме того, образующиеся в избытке -цепи глобина вступают в соединение с SH-груп- пами мембраны эритроцитов и повышают их проницаемость. В результате усиливается гемолиз и развивается неэффективный эритропоэз (значительная часть образовавшихся эритроцитов разрушается уже в костном мозге, в результате чего уменьшается их выход из него. Анемии вследствие нарушения кровообразования ( дизэритропоэтические анемии) По патогенезу нарушения эритропоэза выделяют следующие их разновидности: 1) дисрегуляторная , обусловленная количественным и качественным нарушением синтеза эритропоэтина (снижением продукции их стимуляторов и увеличением образования их ингибиторов), синтеза и активности ферментов, участвующих в образовании порфирина, гема и/или глобина; 2) дефицитная, развивающаяся из-за недостатка в организме неорганических и органических веществ, крайне необходимых для синтеза полноценных эритроцитов; 3) гипопластическая или апластическая, возникающая в результате угнетения красного ростка костного мозга вследствие либо его разрушения, либо его сдавления или замещения другой тканью. ДЕФИЦИТНЫЕ АНЕМИИ К ним относятся: а) железодефицитные (частота их развития составляет 80-95 %); б) В12-дефицитные; в) фолиево-дефицитные; г) белково-дефицит- ные; д) смешанные. Железодефицитные анемии Следует отметить, что в норме из 10-15 мг железа, поступающего в сутки с пищей организмом утилизируется только 1,2-1,5 мг. Этиология. Железодефицитные анемии возникают в результате: 1) снижения поступления железа с пищей;
а) дефицита или отсутствия соляной и других (аскорбиновой, лимонной и др.) кислот, приводящих в норме к превращению (восстановлению) оксидного железа (Fe3+ ) в закисное железо (Fe2+), которое в виде ферритина (как и гемоглобин, и миглобин) у здорового человека хорошо всасывается через слизистую кишечника, б) повреждения микро- и макроворсинок слизистой кишки; 3) расстройств транспорта вновь окисленного в плазме крови железа в комплексе с трансферритином (-глобулином), что наблюдается при гипотрансферритикемии, наличии антител к трансферритину;
Патогенез данной анемии представлен на схеме 23-2. Экзогенный или эндогенный дефицит железа характеризуется снижением его содержания в сыворотке крови, костном мозге и истощением резервов железа, что проявляется исчезновением гемосидерина (темно-желтого железосодержащего пигмента) в макрофагах печени и селезенки, снижением содержания сидеробластов в костномозговой ткани. В результате уменьшается синтез гемоглобина и пролиферативная активность ядерных эритроидных клеток. Дефицит железа отражается также на активности тканевого дыхания вследствие снижения уровня железосодержащих клеточных ферментов (цитохрома С, цитохромоксидазы, сукцинатдегидрогеназы, пероксидазы). Клиническая картина. Развивается гипохромная (ЦП 0,86), микроцитарная анемия. Появляется много патологически измененных форм (пойкилоцитов и анизоцитов). Развивающаяся не только гемическая, но и тканевая гипоксия при железодефицитных анемиях приводит к формированию атрофических и дистрофических процессов в тканях и органах. В наибольшей степени это проявляется в поражении слизистой пищеварительного тракта, тканей миокарда, печени и почек. Признаками дефицита железа в организме являются также специфические сидеропенические симптомы – сухость и вялость кожи, повышенная ломкость ногтей, выпадение волос, разрушение зубов, мышечная слабость, извращение вкуса, ночное недержание мочи. После снижения запасов железа компенсаторно стимулируется как всасывание, так и синтез трансферрина, а также эритропоэз (увеличение числа более мелких эритроидных клеток в результате активизации продукции эритропоэтина). 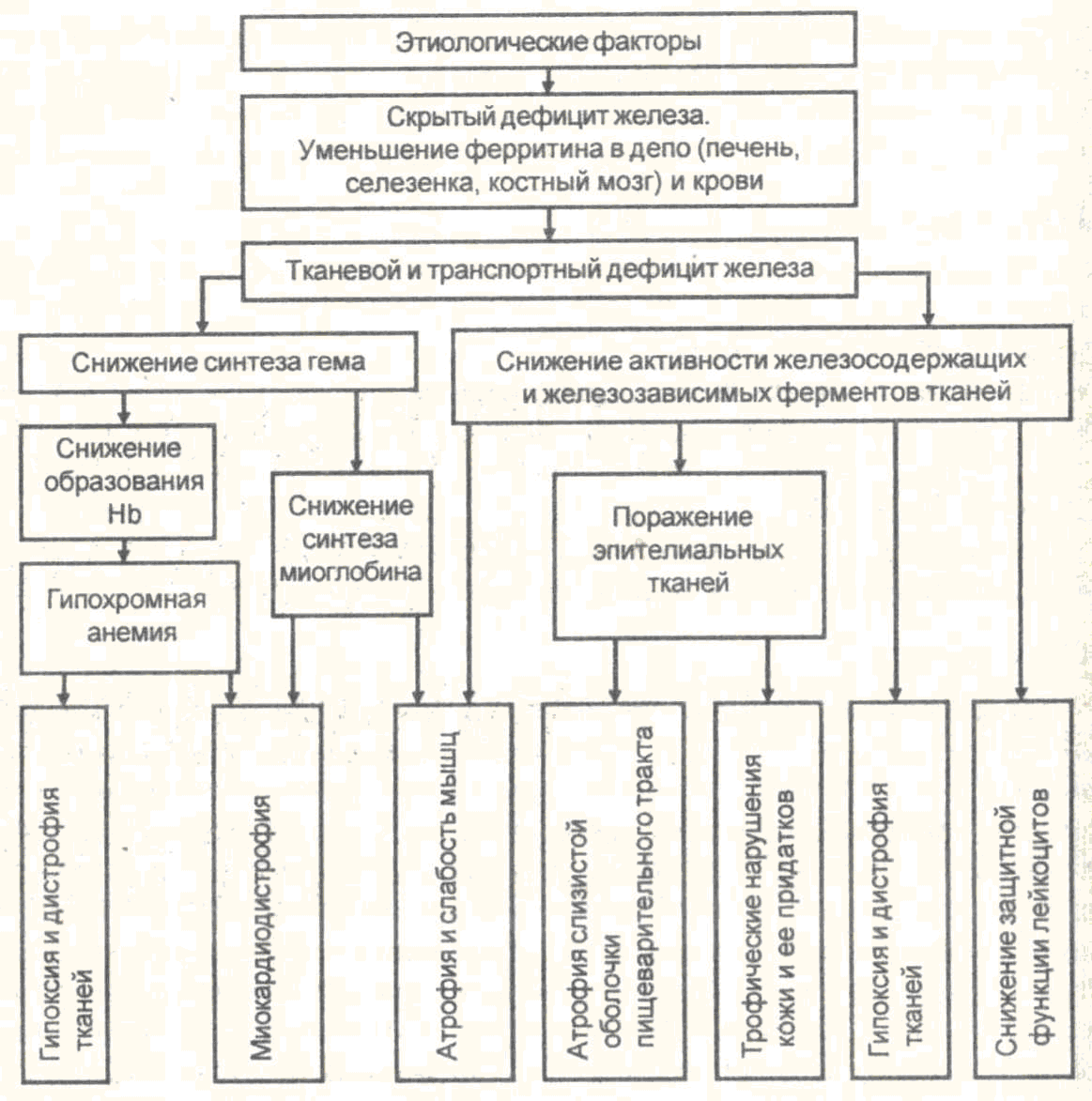 Схема 23-2. Патогенез железодефицитной анемии (Окороков А.Н., 2001) В12- и фолиеводефицитная анемия Дефицит витамина В12 (внутреннего и внешнего фактора Кастла, цианокобаламина) и фолиевой (фолиновой) кислоты приводит к нарушению клеточного деления всех активно пролиферирующих клеток организма. Последнее связано со снижением синтеза пуриновых и пиримидиновых оснований, а следовательно - снижением синтеза нуклеиновых кислот (ДНК, РНК). На уровне системы крови развивается замена нормобластичеческого типа кроветворения мегалобластическим в сочетании с неэффективным миелопоэзом. Этиология. Развитие абсолютного или относительного недостатка витамина В12 и фолиевой кислоты может определяться следующими факторами:
6) повышенная потребность в витамине В12 (быстрый рост организма, беременность, гипертиреоз, новообразования). Патогенез. Дефицит витамина В12 и фолиевой кислоты приводит к снижению активности В12- зависимых энзимов, определяющих превращение фолиевой кислоты в ее коферментную форму тетрагидрофолиевую (фолиновую) кислоту, без которой невозможен синтез тимидина, входящего в состав ДНК. Это в основном и приводит к нарушению клеточного деления активно регенерирующих клеток кроветворной ткани. Развивается мегалобластический тип кроветворения, который характеризуется снижением числа митозов (нормо- бласт дает 3 митоза, мегалобласт – 1 митоз), удлинением времени митотического цикла, более ранним насыщением гемоглобином созревающих клеток эритроидного ряда, внутрикостномозговым разрушением мегалоцитов и укорочением (до 30-40 дней) продолжительности их жизни. Этиология и патогенез В12-дефицитной анемии представлены на схеме 23-3. Клиническая картина. Формируется гиперхромная (ЦП1), мегалоцитарная анемия Аддисона-Бирмера, для которой характерны патологические включения в макроцитах (тельца Жолли, кольца Кебо, базофильная пунктация). При этом отмечается гиперсегментация ядер нейтрофилов и снижение в крови количества ретикулоцитов, лейкоцитов и тромбоцитов. Картина периферической крови представлена на схеме 23-5. 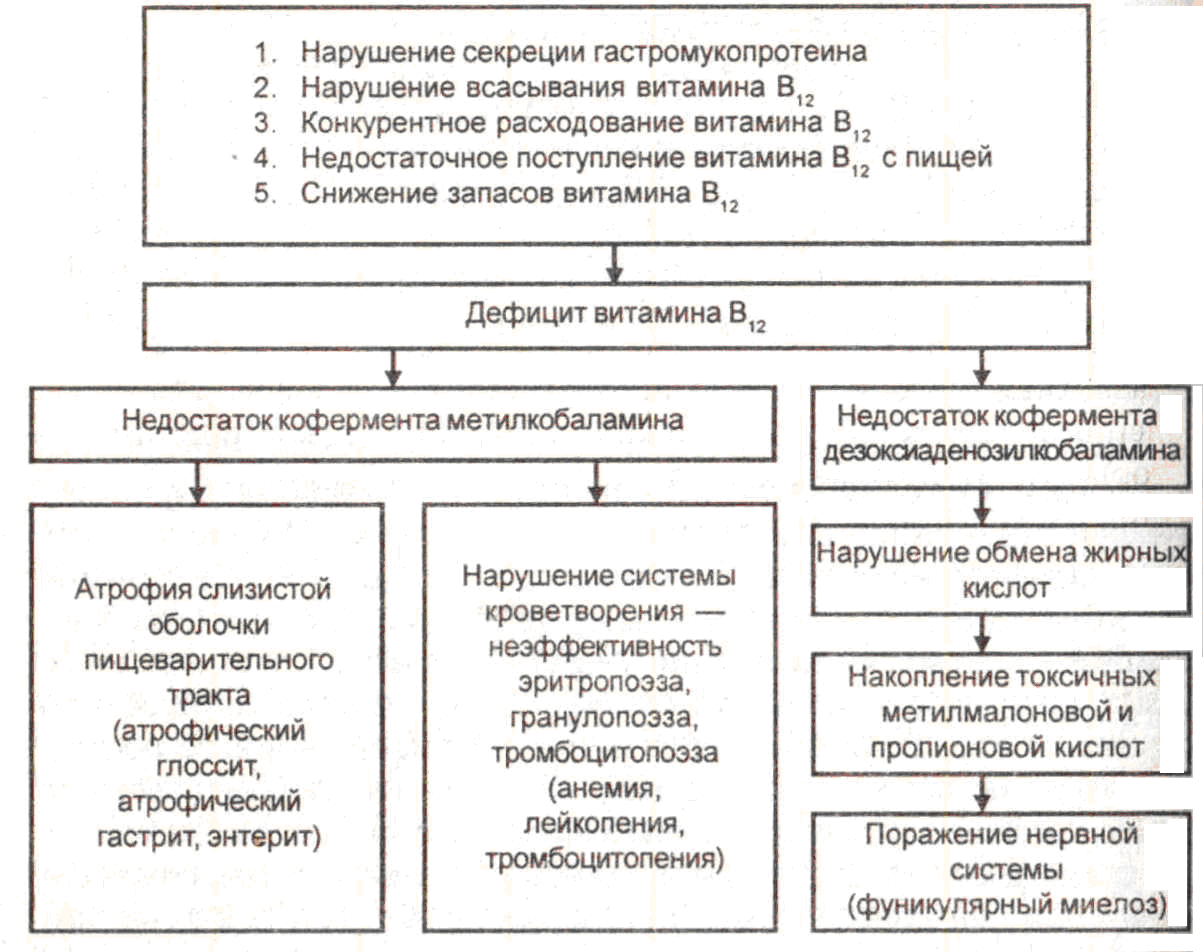 Схема 23-3. Этиология и патогенез В12-дефицитной анемии (Окороков А.Н., 2001) Изменения со стороны системы пищеварения проявляются в виде развития воспалительно-атрофических процессов в связи с нарушением регенерации эпителия слизистой пищевого канала (глоссит, стоматит, эзофагит, ахилический гастрит, энтерит). Патология нервной системы (главным образом в виде дегенерации задних и боковых столбов спинного мозга – фуникулярного миелоза, а также поражений черепно-мозговых и периферических нервов) определяется тем, что при недостатке витамина В12 в тканях накапливаются пропионовая и метилмалоновая кислоты, которые являются токсичными для нервной ткани. При этом развиваются разнообразные неврологические симптомы – парестезии, дефицит моторики, расстройства глубокой чувствительности, болевой синдром и др. Гипо - и апластические анемии Встречаются с частотой 1 на 200000-250000 лиц, независимо от пола. Возникают в результате костномозговой недостаточности, приводящей к уменьшению количества Нb и эритроцитов в крови, а также развитию панцитопении. Выделяют два вида костномозговой недостаточности: 1) абсолютную, при которой костный мозг не способен вырабатывать необходимое количество форменных элементов крови для возмещения их физиологической утраты. В принципе это состояние отмечается тогда, когда кроветворная ткань почти полностью замещается соединительно-тканными структурами, особенно жировой тканью или опухолями; 2) относительную, при которой костный мозг хотя и способен возмещать физиологические потери, тем не менее, продукция эритроцитов становиться меньше потребностей организма (например, при интенсивном гемолизе, острой кровопотере и т.д.) По этиологии различают наследственные и приобретенные гипопластические анемии. Первые чаще бывают общими и возникают в результате генетически обусловленного дефекта развития стволовых и коммитированных клеток. Это приводит к угнетению и эритроилного, и миелоидного и мегакариоцитарного ростков костного мозга. Проявляется развитием анемии Фанкони и семейной анемии (Эстрена-Дамешека). Однако может развиваться наследственное парциальное избирательное поражение эритропоэза с развитием анемии Блекфена-Дайемонда. Вторые развиваются в результате: 1) длительных и интенсивных инфекций (особенно вирусных); 2) интоксикаций, особенно химическими веществами (бензолом, парами ртути, красителями, лекарственными средствами: цитостатиками, антибиотиками, сульфаниламидами, противосудорожными и антитиреоидными препаратами); 3) ионизирующего излучения; 4) развития гипотиреоза, цирроза печени, хронической почечной и надпочечниковой недостаточности; 5) сдавления и замещения эритропоэтической ткани опухолями (доброкачественными и, особенно, злокачественными); 6) дефицита питательных веществ, а также многих витаминов (особенно, В12, фолиевой кислоты и В6), макро- и микроэлементов и т.д. В патогенезе гипо- и апластических анемий важное значение имеют расстройства регуляции эритропоэза и развитие аутоиммунных процессов, сопровождающихся уменьшением количества стволовых клеток или потерей ими и другими предшественницами эритропоэза, эритропоэтинчувствительными клетками, эритробластами, пронормобластами способности к пролиферации и дифференциации, неполноценностью гемопоэтического микроокружения (схема 23-4). На фоне значительного опустошения костного мозга, вызванного значительным замещением его его жировой или опухолевой (миело-, лимфолейкозной, миеломной, гематосаркомной) тканью обнаруживаются лишь небольшие очаги кроветворения. Схема 23-4. Патогенез апластической анемии (Окороков А.Н., 2001) Основными клиническими проявлениями гипо- и апластических анемий являются снижение в крови количества как эритроцитов, ретикулоцитов и гемоглобина, так и лейкоцитов (особенно нейтрофилов) и тромбоцитов, а также повышение СОЭ. Количество ретикулоцитов чаще в пределах нормы, а уровень эритропоэтина, как правило, увеличен. На фоне прогрессирующей тромбоцитопении развивается геморрагический синдром. 23.3. Основные принципы терапии анемий Выделяют этиотропный, патогенетический (заместительный, стимулирующий, тормозный, корригирующий) и симптоматический принципы лечения различных видов анемий. |