ТЭЛА. Тромбоэмболия легочной артерии (тэла)
 Скачать 4.9 Mb. Скачать 4.9 Mb.
|

Под шоком понимают остро возникшую несостоятельность кровообращения с критическим расстройством тканевой перфузии, которое ведёт к дефициту кислорода в тканях, повреждению клеток и нарушению функции органов [92]. Отмечается нарушение гемодинамики с артериальной гипотонией в результате дисфункции сердца, что требует назначения вазопрессорных препаратов. Развитие шока у больных ТЭЛА отражает недостаточность компенсаторных механизмов поддержания АД и/или перфузии тканей, что сопровождается повышением летальности в 3-7 раз. Таким образом, развитие шока позволяет быстро оценить прогноз для жизни (см. раздел « стратификация риска и прогноз », таблицу 4) [20].
Вирхов первым обратил внимание на то, что лёгочная эмболия может приводить к развитию инфаркта лёгкого, определяемого как некроз ткани лёгкого дистальнее области эмболической обструкции. Внезапная тромботическая окклюзия сосудов сопровождается повышением давления проксимальнее тромба и снижением или полным прекращением кровотока дистальнее. Это приводит к целому ряду последствий, включая уменьшение в альвеолах количества сурфактанта, повышение содержания белка и высвобождение провоспалительных медиаторов [20]. Инфаркт лёгкого развивается, если дистальные эмболии вызывают полную закупорку ветви ЛА малого калибра, в то же время проксимальная эмболия редко приводит к инфаркту. Последнее обстоятельство обусловлено тем, что паренхима лёгких обеспечена четырьмя источниками поступления кислорода: воздухоносные пути, лёгочные артерии, коллатеральные пути через бронхиальные артерии и обратная диффузия из венозного русла лёгких. Эти механизмы компенсации могут не функционировать у больных с патологией лёгких или имеющейся ко времени развития ТЭЛА сердечной недостаточностью. Такая ситуация предполагает даже при проксимальной эмболии развитие инфаркта лёгкого [62]. Инфаркт лёгкого у здоровых до развития ТЭЛА пациентов развивается в 10%, однако если в эмболизированной области исходно были нарушения оксигенации (заболевания лёгких или застой по малому кругу), частота инфаркта возрастает до 30% [31]. В случае развития инфаркта лёгкого через неделю отмечается полная некротизация ткани, которая отграничивается перифокальной инфильтрацией. Период рассасывания погибших масс продолжается две недели. Затем на их месте формируется пневмосклероз. В целом весь процесс, включая заключительную стадию (формирование пневмосклероза), занимает от двух до трёх месяцев. Инфаркт лёгкого может осложниться присоединением пневмонии , абсцессом, гангренизацией. При бронхогенном проникновении инфекции в зоне инфаркта или вокруг него развивается воспалительный процесс (инфаркт-пневмония), который может сопровождаться распадом лёгочной ткани. В таком случае в течение нескольких дней вследствие секвестрации некротического очага образуется полость значительных размеров [62].
Эмболическая окклюзия лёгочной артерии не обязательно ведёт к развитию инфаркта лёгкого. На самом деле, в отсутствие фонового заболевания сердца или лёгких истинный инфаркт (то есть некроз стенки альвеол) развивается не так уж часто. Неполный инфаркт лёгких впервые описали Hampton и Castleman, определив его как внутриальвеолярное кровоизлияние без некроза стенки альвеол [20]. Кровоизлияние происходит из-за градиента между низким давлением в лёгочных артериолах дистальнее эмбола и нормальным – в концевых ветвях бронхиальных артерий [64]. Неполный инфаркт развивается обычно у больных без сопутствующей патологии и разрешается в течение 1 недели, в то время как рентгенографические признаки полного инфаркта лёгких (с некрозом ткани) сохраняются более 2 недель (при формировании постинфарктного пневмосклероза – всю жизнь). Кровоизлияние в альвеолы без развития некроза – наиболее частое событие в первые двое суток ТЭЛА. Рентгенографические признаки этого поражения полностью исчезают в течение 2-4 дней, что соответствует рассасыванию внутриальвеолярных кровоизлияний по секционным данным. При персистировании полной окклюзии обычно через 2 дня развивается некроз альвеолярной стенки (полный инфаркт лёгкого). Важность неполного инфаркта заключается в том, что он увеличивает вероятность постановки правильного диагноза, так как определяется при использовании визуализационных методик, таких как спиральная КТ или трансторакальная сонография [20]. Кровоизлияние при полном или неполном инфаркте лёгкого лежит в основе появления такого симптома ТЭЛА, как кровохарканье.
Сразу после тромботической окклюзии ветви лёгочной артерии развивается бронхоконстрикция. В секционных исследованиях показано, что обструкции подвергаются в основном мелкие бронхи. Предполагается, что снижение объёма лёгких, уменьшение статической и динамической податливости у больных с ТЭЛА происходит вследствие констрикции мелких дыхательных путей. Принимая во внимание, что бронхоспазм можно предотвратить, назначив предварительно гепарин и блокаторы серотонина, можно считать, что уменьшение диаметра дыхательных путей, по крайней мере отчасти, связано с высвобождением бронхоконстрикторных медиаторов из тромбоцитов и воспалительных клеток (ФАТ, тромбоксан А 2 и др. - см. « роль воспаления в патогенезе ВТЭ »). Кроме того, другие медиаторы, высвобождаемые при ТЭЛА, такие как фактор активации тромбоцитов или тромбоксан, могут способствовать бронхоконстрикции [20]. Бронхоспазм может обусловить появление свистящих хрипов при аускультации больного ТЭЛА.
Изредка при ТЭЛА рентгенографически определяются признаки отёка лёгких. Однако даже при использовании спиральной КТ, признаки отёка лёгких (гетерогенные области изменений по типу матового стекла, локализующиеся сегментарно или субсегментарно) выявляются менее чем в 10% случаев. Он развивается в результате повышения давления в лёгочных капиллярах интактных областей, а также увеличения проницаемости альволярно-капиллярной мембраны. Предполагается, что развитие отёка лёгких вносит вклад в нарушение вентиляционно-перфузионных отношений [20].
При хронической ТЭЛА отёк лёгких развивается чаще. В этом случае на КТ определяются участки повышенной плотности по типу матового стекла, чётко отграниченные от участков повышенной прозрачности, расположенных дистальнее окклюзированных артерий и, соответственно, лишенных притока крови. Наличие изменений по типу матового стекла тесным образом связано с расширением лёгочных артерий, что отмечается в 70% случаев при ТЭЛА. Таким образом, участки затемнения имеют смешанный генез: повышенная перфузия (гиперемия) и накопление жидкости в ткани перфузируемых областей. Тесное соседство участков уплотнения и гипоперфузии создаёт хорошо узнаваемую картину «мозаичной олигемии» [20].
Реперфузионный отёк лёгких представляет собой острый некардиогенный отёк, который развивается в 90-100% случаев после тромбэктомии по поводу массивной ТЭЛА или тромбоэндариэктомии в связи с формированием стенозов при хронической посттромбоэмболической лёгочной гипертензии. На рентгенограммах грудной клетки в течение первых 2 дней после операции выявляются гетерогенные участки уплотнения лёгочной ткани, главным образом дистальнее реканализованного сосуда. Реперфузионный отёк лёгких может развиваться также после частичного или полного тромболизиса. Основным патофизиологическим механизмом данного расстройства является быстрое повышение кровотока и кровяного давления в областях дистальнее реканализованной лёгочной артерии. Определённый вклад вносят и другие факторы: механический стресс вследствие хирургического вмешательства и биохоимические изменения (например, высвобождение радикалов кислорода нейтрофилами, нарушение синтеза сурфактанта) [20].
Сурфактант представляет собой смесь фосфолипидов и белков, которая выстилает внутреннюю поверхность альвеол. Сурфактант снижает поверхностное натяжение и защищает альвеолы от спадения, особенно в конце выдоха. Сурфактант также способен снижать транссудацию жидкости в интерстициальное и альвеолярное пространство. Таким образом, внезапная закупорка лёгочной артерии тромбом приводит к повреждению системы сурфактанта. В результате развиваются ателектаз, транссудация жидкости и клеток в повреждённую ткань лёгких. Вначале ТЭЛА приводит к повышению общего содержания фосфолипидов. Причина такого повышения неизвестна; она может быть связана или с повышенной продукцией или с повышенной секрецией из поврежденных клеточных мембран или с нарушением обратного захвата фосфолипидов альвеолоцитами II типа или макрофагами, подобно процессам при лёгочном альвеолярном протеинозе. Кроме того, гипервентиляция, характерный симптом ТЭЛА, может усиливать продукцию фосфолипидов через холинергический и/или бета-адренергический механизмы. Однако качественные изменения сурфактанта, состоящие в снижении содержания главным образом фосфатидилхолина и фосфатидилглицерола, могут свидетельствовать в пользу повреждения альвеолоцитов II типа, как основного патофизиологического механизма исходного повышения общего уровня фосфолипидов. По прошествии острой стадии ТЭЛА, общее содержание фосфолипидов в лёгких снижается, что опять же может отражать снижение секреторной способности повреждённых альвеолоцитов II типа. Другой возможный механизм снижения концентрации фосфолипидов — рассасывание тромба с последующей реперфузией ткани лёгкого [20].
Ателектазы вследствие ТЭЛА — частая рентгенографическая находка, особенно при одновременном развитии инфаркта лёгкого. Причиной ателектазов являются как уменьшение количества сурфактанта, так и кровоизлияние в альвеолы. Указанные изменения могут запускаться эмболической обструкцией лёгочного сосуда, кровоснабжающего соответствующую часть лёгочной паренхимы, а также феноменом «воздушного шунта», когда регионарная гипокапния вследствие гипоперфузии приводит к бронхоспазму, что поддерживается высвобождаемыми из тромбоцитов медиаторами [20].
Нарушения газообмена — одно из ключевых звеньев патогенеза ТЭЛА. Типично развитие гипоксемии и гипокапнии. Характер нарушений прямо или косвенно зависит от следующих факторов: размер и состав тромба; протяжённость окклюзии; фоновое состояние сердечно-сосудистой и дыхательной систем; время, прошедшее с момента эмболии. Наиболее частый признак ТЭЛА — падение парциального напряжения кислорода в артериальной крови, что наблюдается уже при окклюзии 13% лёгочного сосудистого русла. Развитие гипоксемии связано с увеличением альвеолярного мёртвого пространства, право-левым сбросом крови, вентиляционно-перфузионным несоответствием и с низким уровнем кислорода в смешанной венозной крови. Нарушение вентиляционно-перфузионных отношений развивается вследствие перераспределения кровотока в обход эмболизированных областей, что приводит к повышенной перфузии неэмболизированных участков лёгких, в том числе плохо вентилируемых (с исходно низким V/Q). Кроме того, ателектазы лёгких, образовавшиеся дистальнее обтурированного сосуда, сохраняются даже после растворения тромба и реперфузии (опять же перфузия невентилируемых областей) [20]. Этот процесс может стать особенно значимым спустя несколько дней после ТЭЛА. Повышение давления в правом предсердии при наличии открытого овального окна (у 25% здоровых не закрыто, но в обычных условиях не функционирует, так как прикрыто особой заслонкой со стороны левого предсердия) сопровождается право-левым сбросом на уровне предсердий [20, 31, 62]. Эти состояния следует заподозрить, если гипоксемия выражена сильнее, чем можно было ожидать, исходя из других клинческих признаков, а также при сопутствующей гиперкапнии и отсутствии эффекта от кислородотерапии [31]. Постэмболический отёк лёгких вносит свою лепту в развитие артериальной гипоксемии. Наконец, у больных массивной ТЭЛА с циркуляторным коллапсом, увеличение сердечного выброса в результате лечебных мероприятий может привести к падения PaO 2 вследствие усиления физиологического шунта через плохо вентилируемые области (с исходно низким вентиляционно-префузионным отношением) [20]. Ещё одним фактором, способствующим развитию артериальной гипоксемии, является чрезвычайно низкая сатурация смешанной венозной крови вследствие сниженного сердечного выброса: такая кровь не успевает полностью насытиться кислородом, проходя через альвеолярные капилляры областей лёгких с повышенной перфузией [31]. В то же время, гипоксемия при ТЭЛА не может быть объяснена единственно механической обструкцией сосудистого ложа и шунтированием крови. Учитывая, что тяжёлые нарушения газоомбена отмечаются даже у пациентов с небольшими эмболиями, гипоксемия скорее всего связана ещё и с высвобождением медиаторов воспаления (см. « роль воспаления в патогенезе ВТЭ »), снижающих активность сурфактанта, изменяющих проницаемость сосудов и функциональные внутрилёгочные шунты [20]. Хотя при ТЭЛА элименация CO 2 затруднена, гиперкапния развивается редко вследствие компенсаторной гипервентиляции. Достаточная же для развития гиперкапнии степень обструкции сосудистого русла лёгких, приводит, как правило, к фатальной правожелудочковой недостаточности [31]. Скорее, наоборот: для ТЭЛА характерно развитие артериальной гипокапнии и алкалоза , хотя гипокапния встречается не так часто, как гипоксемия. Возможно, гипокапния связана с частым поверхностным дыханием, наблюдаемым при ТЭЛА. Учитывая, что гипоксемия редко достигает такой выраженности, чтобы объяснить гипервентиляцию активацией хеморецепторов, нужно думать о существовании других видов афферентной импульсации в дыхательный центр. Лежащий в основе такого типа дыхания механизм неизвестен, но, возможно, связан с активацией С-волокон (J-рецепторов) и ирритантных рецепторов при эмболической окклюзии [20].
Предполагается, что плевральные боли при периферической ТЭЛА обусловлены воспалительным ответом париетальной плевры, примыкающей к поражённой области лёгких. Вовлечение плевры изначально приводит к локальному накоплению жидкости, в результате чего может развиться базальный плевральный выпот. Как правило, жидкость скапливается сразу после ТЭЛА. Однако выпот обычно небольшой и односторонний, достигает максимальной выраженности в течение первых 3 дней. Плевральный выпот определяется в 51% случаев ТЭЛА при рентгенографическом исследовании, в 57% - при КТ и в 70% - при трансторакальном УЗИ. Однако частота плеврального выпота при ТЭЛА недооценивается: объём выпота зачастую настолько мал, что не распознаётся при обычной рентгенографии грудной клетки, а порой, даже не подумав о возможности ТЭЛА, больного не обследуют. Согласно современным представлениям в патогенезе плеврального выпота участвуют два основных механизма. Первый — повышение проницаемости сосудов лёгочной паренхимы. Около 20% жидкости поступает в интерстиций. Как только объём жидкости превышает способность лимфатических сосудов дренировать её, развивается плевральный выпот. Это может быть вызвано ишемией ткани и/или высвобождением провоспалительных медиаторов. Второй механизм связан с повышением капиллярного давления в париетальной плевре. В связи с тем, что лимфа в конце концов впадает в венозную систему, повышение системного венозного давления препятствует лимфатической системе дренировать плевральную полость, в результате чего в ней скапливается жидкость. Анализ плеврального выпота не позволяет сделать диагностического заключения, так как при ТЭЛА выпот может иметь характер как транссудата , так и экссудата , а явно геморрагическим бывает лишь в небольшом проценте случаев. Плевральный выпот при ТЭЛА богат как клеточными элементами, так и биомолекулами. В некоторых случаях результаты анализа плеврального ТЭЛА-ассоциированного выпота соответствуют диагнозам «эмпиема» или «хилоторакс», чем в очередной раз подтверждается, что в образовании выпота в зависимости от конкретных респираторных и гемодинамических нарушений участвуют разные механизмы. Макроскопически он может быть прозрачным, жёлтыми, слегка подкрашенным кровью или геморрагическим. Когда отмечается очень высокий цитоз, концентрация глюкозы и pH бывают низкими. Среди лейкоцитов встречаются как нейтрофилы, так и мононуклеары. Также может повышаться содержание мезотелиальных клеток и эозинофилов (более 10% всех лейкоцитов). В общем, ТЭЛА является причиной 4% эозинофильных плевральных выпотов [20].
Тромбоз запускает воспалительно-прокоагулянтный каскад, что приводит к повреждению венозной стенки. Процесс рассасывания тромба аналогичен таковому при заживлении ран: лейкоцитарная инфильтрация с возможным исходом в фиброз венозной стенки. Медиаторами этого процесса являются провоспалительные цитокины, такие как ФНО-α, ИЛ-1β (из клеток повреждённой вены и местных лейкоцитов), которые способствуют дальнейшей активации лейкоцитов, стимулируют высвобождение хемотаксических факторов, усиливают тромбоз и повышают экспрессию клеточных молекул адгезии. Кроме того, тромбин действует как провоспалительный медиатор, который не только усиливает коагуляционный каскад, но также стимулирует прокоагулянтный потенциал эндотелиальных клеток с повышением экспрессии молекул клеточной адгезии и высвобождением медиаторов. Молекулы клеточной адгезии, Р- и Е- селектины и ICAM-1 – ключевые факторы, способствующие адгезии и миграции лейкоцитов соответственно. Эмиграция лейкоцитов в сосудистую стенку сопровождается высвобождением цитотоксических воспалительных медиаторов, которые повреждают сосуд. Воспалительный процесс напрямую задействован в патогенезе ТЭЛА. Так, повышение проницаемости лёгочных капилляров является следствием воздействия воспалительных медиаторов. Воспаление также играет роль в нарушении газообмена и в развитии бронхоспазма.
В нескольких исследованиях показано, что после ТЭЛА поражённые области инфильтрируются воспалительными клетками. Микроэмболия, индуцированная внутривенным введением тромбина, приводит к притоку нейтрофилов в лёгкие в течение 15 минут. Повышенное поступление нейтрофилов в лёгочную циркуляцию сопровождалось снижением общего цитоза крови. Лейкоциты могут аккумулироваться интраальвеолярно, а также инфильтрировать стенку лёгочных сосудов. Взаимосвязь между венозным тромбозом и воспалением стенки вен (флебитом) была подтверждена в эксперименте на грызунах, когда тромбоз индуцировали венозным стазом. Позже клеточную инфильтрацию подтвердили в нескольких исследованиях на животных при экспериментальной ТЭЛА. У крыс в подобных исследованиях отмечалась выраженная нейтрофильная инфильтрация стенки лёгочной артерии. Она сохраняется более 8 дней после ТЭЛА, достигая максимума на 2-е сутки. Макрофагальная инфильтрация стенки лёгочной артерии максимальна в первые сутки. В другом исследовании на крысах при экспериментальном тромбозе нижней полой вены определяли особенности воспаления по гистопатологии и морфометрии лейкоцитов. Взятые вместе, эти исследования показывают, что тромбоз приводит к ранней (1-й день) инфильтрации венозной стенки нейтрофилами, затем моноцитами/макрофагами и лимфоцитами (на 3 и 6-е сутки).
Эксперименты как in vivo, так и in vitro продемонстрировали, что компоненты воспалительного и коагуляционного каскада взаимосвязаны. Например, тромбин играет центральную роль в свёртывании и воспалении. Эта сериновая протеаза (тромбин), образующаяся из протромбина при активации коагуляционного каскада, является важным стимулятором агрегации тромбоцитов и превращает фибриноген в фибрин (см. каскад свёртывания рис.4 ), контролируя конечный этап тромбообразования. Тромбин также выполняет функции воспалительного медиатора через взаимодействие со специфическими клеточными рецепторами. Тромбин стимулирует экспрессию P-селектина (CD62E), а также продукцию простациклина (PGI 2 ), ФАТ и хемокинов эндотелиальными клетками. Он также активирует гладкомышечные клетки, моноциты и полиморфноядерные лимфоциты.
В недавнем исследовании состава жидкости бронхоальвеолярного лаважа (БАЛ) показано повышение уровня белка у больных ТЭЛА, чем подтверждается нарушение альвеолокапиллярной проницаемости. Повышение проницаемости, в свою очередь, по-видимому, связано с высвобождением множества воспалительных медиаторов, таких как фактор активации тромбоцитов (ФАТ) и другие производные арахидоновой кислоты. Вслед за развитием ТЭЛА, концентрация ФАТ значительно повышается в жидкости БАЛ. Фактор активации тромбоцитов — важный фосфолипидный медиатор воспалительных и аллергических реакций, участвует в острых, равно как в продолжительных физиологических и воспалительных лёгочных процессах. Клетки (эндотелиальные, эпителиальные, нейтрофилы, альвеолярные макрофаги) могут продуцировать фактор активации тромбоцитов при локальной гипоксемии. Снижение уровня фосфатидилглицерола, обладающего анти-ФАТ активностью, способствует усилению действия ФАТ в альвеолах. Фактор активации тромбоцитов обладает плейотропной активностью. Он может участвовать в патогенезе гипоксемии, лёгочной гипертензии и повышения сопротивления дыхательных путей. В частности, ФАТ может способствовать развитию интерстициального отёка через повышение сосудистой проницаемости, стимуляцию синтеза ряда медиаторов воспаления, вызывая бронхоконстрикцию и провоцируя сокращение лёгочных вен, что влечёт за собой повышение давления в лёгочных капиллярах. Более того, ФАТ стимулирует приток нейтрофилов и эозинофилов в лёгкие и в плевральный выпот. Наконец, ФАТ обладает способностью обеспечивать как прайминг, так и активацию клеток. У некоторых больных ТЭЛА в жидкости БАЛ определяется повышенный уровень ФАТ-ацетилгидролазы. Этот фермент модулирует концентрацию провоспалительных медиаторов, ФАТ и ФАТ-подобных продуктов окисления фосфолипидов в тканях и биологических жидкостях. ФАТ-ацетилгидролаза отчасти поступает из плазмы через альвеоло-капиллярную мембрану, отчасти высвобождается инфильтрирующими воспалительными клетками, такими как нейтрофилы. Альвеолоциты II типа и макрофаги — также потенциальные источники ФАТ-ацетилгидролазы в жидкости БАЛ. При ТЭЛА могут определяться и другие синтезируемые de novo медиаторы. В экспериментах на животных микроэмболии лёгочной артерии, индуцированные тромбином, сопровождались повышением уровней LT-B4, моногидроксиэйкозотетраеновой кислоты (5-HETE и 15-HETE) в лимфе лёгких. Эти продукты липоксигеназного пути увеличивают продукцию радикалов кислорода полиморфноядерными нейтрофилами, в результате чего повышается сосудистая проницаемость. У кроликов ТЭЛА ассоциировалась со значительным транзиторным повышением уровня тромбоксана А 2 , в то время как уровень PGI 2 оставался неизменным. Тромбоксан А2 влияет на сосудистую проницаемость, а также вызывает бронхоконстрикцию.
Исследовалась роль многих цитокинов, потенциально вовлечённых в патогенез ТЭЛА (см. факторы риска ВТЭ ). Например, экспрессия хемотаксического белка моноцитов-1 (MCP-1) в стенке поражённой лёгочной артерии повышалась в первые 3 дня ТЭЛА. Этот хемокин оказывает сильное хемотаксическое действие на моноциты/макрофаги и участвует в ремоделировании лёгочной артерии. После венозного тромбоза более 6 дней отмечается повышение уровня ENA-78 (эпителиально-нейтрофильный активирующий белок 78), ФНО-α, ИЛ-6 и MCP-1, в то время как макрофагальный воспалительный белок-1 (MIP-1) достигает максимума к третьему дню после тромбоза. Кроме того, в организующихся тромбах в эксперименте на крысах отмечается экспрессия VEGF (сосудистого эндотелиального фактора роста) и основного фактора роста фибробластов (bFGF), чем подтверждается их участие в разрешении тромботического процесса. Иммуногистохимически определена локализация VEGF на моноцитах, эндотелиальных клетках и веретеновидных клетках в тромбах. Их обнаруживали во внеклеточном матриксе. Усиление экспрессии ангиогенных факторов роста может ускорять реканализацию тромбов, таким образом, уменьшая частоту поздних осложнений. Концентрация VEGF повышалась и в плевральном выпоте при ТЭЛА. Известный также как фактор сосудистой проницаемости или васкулотропин, VEGF представляет собой цитокин плейотропного действия, который в дополнение к своим ангиогенным свойствам, повышает сосудистую проницаемость. В ряде случаев выпот в плевральную полость может быть связан именно с повышением его концентрации. Источник VEGF при ТЭЛА неизвестен, однако, возможно, он происходит из тромба самого по себе, ибо выделяется и при агрегации и при активации тромбоцитов. Для установления роли различных цитокинов в патогенезе воспаления при ТЭЛА в эксперименте на крысах вводили антитела к различным цитокинам. Показано, что анти-ФНО лучше других предотвращали инфильтрацию венозной стенки нейтрофилами после индукции тромбоза. Кроме того, анти-ICAM-1 и анти-ФНО подавляли выход моноцитов/макрофагов. Эти данные подтверждают, что тромбоз вен связан со значительным воспалением венозной стенки и что воспалительные медиаторы и цитокины играют роль в процессах, следующих за ТЭЛА. Некоторые цитокины оказывают противовоспалительный эффект и влияют на процессы фибринолиза. Так, нейтрализация противовоспалительного цитокина ИЛ-10 приводит к усилению локального воспаления, в то время как введение человеческого ИЛ-10 дозозависимо уменьшает не только перитромботическое воспаление, но и размер тромба. На другой модели введение провоспалительного и ангиогенного цитокина ИЛ-8 усиливало рассасывание тромба.
Тромбин повышает экспрессию клеточных молекул адгезии. В ряде исследований, кроме того, показано, что тромбоэмболический процесс связан с экспрессией клеточных молекул адгезии, таких как P-селектин, ICAM-1 (CD54) и VCAM-1 (CD106). В экспериментах на животных введение рекомбинантного гликопротеинового лиганда P-селектина приводило к подавлению тромбоза и уменьшало уровень ИЛ-8, тромбоцитарного фактора IV и MCP-1 в венозной стенке, не влияя при этом на лейкоцитарную инфильтрацию стенки сосуда. Уменьшалась выраженность её повреждения, снижался уровень D-димера. Таким образом, молекулы адгезии участвуют как в развитии тромбоза, так и в последствиях ТЭЛА. Блокада молекул адгезии может представлять собой перспективный подход в лечении ВТЭ.
Первые данные относительно естественного течения ВТЭ были опубликованы в 1960-х годах, в основу их легли материалы отделений ортопедической хирургии. Было показано, что ВТЭ начинается с тромбоза глубоких вен (ТГВ) уже во время оперативного вмешательства в 30% случаев. У трети больных в течение нескольких дней тромбы спонтанно лизируются, ещё в 40% они не прогрессируют. Однако у 25% больных образовавшиеся тромбы распространяются в проксимальные вены и обусловливают развитие ТЭЛА. С тех пор представления о естественном течении ВТЭ изменились. Показано, что в общей хирургии ТГВ формируется реже, чем при ортопедических операциях. Риск его развития наиболее высок в первые две недели после оперативного вмешательства, хотя сохраняется в течение 2-3 месяцев. Антитромботическая профилактика значительно снижает частоту периоперационного ВТЭ: чем дольше проводится профилактика, тем меньше риск. Хотя основные данные по естественному течению ТЭЛА получены при изучении материалов хирургических отделений, не следует забывать, что от 50 до 70% клинически явных эпизодов ВТЭ и 70-80% летальных ТЭЛА приходится на нехирургических больных [96]. Преобладают бессимптомные формы заболевания. H. Bounameaux (1999 г.) считает, что на 1000 венозных тромбозов только 100 имеют какие-либо клинические проявления; из них у 60 пациентов разовьется ТЭЛА, но только в 10 случаях она будет иметь клинические признаки) [46]. Клинически манифестное течение ТГВ обусловлено обычно проксимальными тромбами. В 40-50% таких случаев развивается ТЭЛА, часто бессимптомная. Бессимптомная ТЭЛА вообще не редкость в послеоперационном периоде, особенно при бессимптомном течении ТГВ у больных, которым не проводится профилактика. ТЭЛА развивается через 3-7 дней после начала ТГВ. В 10% ТЭЛА заканчивается летально в течение часа от появления симптомов, в большинстве таких случаев клинический диагноз устанавливается неверно. Более 90% всех смертельных исходов приходится на больных, не получавших лечения. Нередко летальной ТЭЛА предшествуют «малые» эпизоды ТЭЛА, которые остаются нераспознанными. В целом анатомически большие эмболы представляют большую опасность, чем маленькие. В редких случаях, тем не менее, эмболия мелких периферических ветвей лёгочной артерии при проходимом главном русле может проявиться тяжёлой симптоматикой и даже привести к внезапной смерти. Любую ТЭЛА следует рассматривать как потенциально опасную, поскольку при этом состоянии возможно рецидивирование, какой бы ни была клиническая тяжесть первого эпизода. Шок или гипотония в качестве первых признаков ТЭЛА отмечается в 5-10%. В 50% случаев больные остаются гемодинамически стабильны, однако отмечаются признаки дисфункции или повреждения правого желудочка, что ухудшает прогноз . В подострой фазе ТЭЛА прогноз в значительной степени зависит от возможности лизиса сгустка и восстановления кровотока в лёгочной артерии и системе глубоких вен. Это определяется рядом факторов, таких как существующая сопутствующая тромбофилия, адекватность антикоагулянтной терапии, наличие постоянных факторов риска. Полное растворение тромбоэмболов в лёгочной артерии отмечается у 2/3 больных. У некоторых пациентов, обследовавшихся по поводу одышки или хронической правожелудочковой недостаточности, была обнаружена тяжёлая лёгочная гипертензия, обусловленная бессимптомными повторными ТЭЛА. Такое течение хронической посстромбоэмболической лёгочной гипертензии отчётливо прослеживается после острой ТЭЛА у части больных и без лечения обычно приводит к смертельному исходу в течение 2-3 лет с момента первичной диагностики. При лечении ТЭЛА хроническая посттромбоэмболическая лёгочная гипертензия формируется в 0.5-5%. Существует значительный риск повторной ТЭЛА, особенно в течение 4-6 недель. Этот риск значительно увеличивается при отсутствии антикоагулянтной терапии. Если не проводится антикоагулянтная профилактика, риск рецидива ВТЭ в течение 3 месяцев составляет 50%. Среди больных, принимающих профилактические дозы антикоагулянтов в течение 3-12 месяцев, при наблюдении в течение 4.5 лет риск фатальной ТЭЛА составлял ежегодно 0.2-0.4%. Частота рецидивирования ВТЭ не зависит от исходной его манифестации (ТГВ или ТЭЛА). При идиопатической форме распространённость рецидивов выше. Риск летальной ТЭЛА выше в случаях, когда первый эпизод ВТЭ проявился изолированным ТГВ [26, 28, 38, 41, 52]. КЛИНИКА И ОСЛОЖНЕНИЯ Лёгочные эмболии даже весьма опытными клиницистами просматриваются более чем в половине случаев. Частично это объясняется скрытым течением тех патологических процессов, которые обычно являются основными источниками лёгочных эмболий (ТГВ, внутриполостные тромбы правого сердца) [65]. Трудности правильной прижизненной диагностики ТЭЛА обусловлены также многообразием клинических проявлений этого заболевания [67].
Источником эмболов при ТЭЛА почти всегда являются вены, особенно нижних конечностей. Заподозрить ТГВ можно при наличии следующей клинической симптоматики [51, 62, 63]:
Однако в половине случаев флеботромбозы бессимптомны [64]. Развитие клинической картины ТГВ зависит от протяжённости тромбоза, степени окклюзии и наличия воспаления. Большинство клинически явных венозных тромбозов начинается с тромбоза глубоких вен голени, но клиническая картина появляется только при распространении тромбоза на проксимальные вены [63]. В качестве причины бессимптомности флеботромбоза прежде всего следует отметить возможность неполной обтурации тромбированной вены. Другой причиной может быть несущественное влияние тромбированной вены на отток крови, например, при изолированном тромбозе глубокой вены берда. Наконец, возможно тромбирование одной из удвоенных поверхностных бедренных или подколенных вен (рисунок 6) [64]. Р 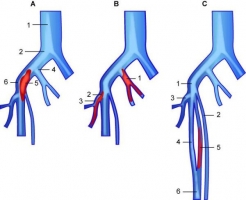 исунок 6. Причины бессимптомности течения ТГВ. A. Бессимптомный тромбоз бедренной и наружной подкожной вен: 1 – нижняя полая вена; 2 – правая общая подвздошная вена; 3 – наружная подвздошная вена; 4 – внутренняя подвздошная вена; 5 – тромб, не обтурирующий просвета; 6 – бедренная вена. B. Бессимптомный тромбоз внутренней подвздошной и глубокой вены бедра: 1 – тромб в просвете правой внутренней подвздошной вены; 2 – тромб, флотирующий в просвете глубокой вены правого бедра; 3 – глубокая вена правого бедра. C. Бессимптомный тромбоз одной из удвоенных бедренных вен: 1 – наружная подвздошная вена; 2 – большая подкожная вена; 3 – глубокая вена бедра; 4 – одна из удвоенных бедренных вен; 5 – тромб в просвете одной из удвоенных бедренных вен; 6 – подколенная вена [64]. исунок 6. Причины бессимптомности течения ТГВ. A. Бессимптомный тромбоз бедренной и наружной подкожной вен: 1 – нижняя полая вена; 2 – правая общая подвздошная вена; 3 – наружная подвздошная вена; 4 – внутренняя подвздошная вена; 5 – тромб, не обтурирующий просвета; 6 – бедренная вена. B. Бессимптомный тромбоз внутренней подвздошной и глубокой вены бедра: 1 – тромб в просвете правой внутренней подвздошной вены; 2 – тромб, флотирующий в просвете глубокой вены правого бедра; 3 – глубокая вена правого бедра. C. Бессимптомный тромбоз одной из удвоенных бедренных вен: 1 – наружная подвздошная вена; 2 – большая подкожная вена; 3 – глубокая вена бедра; 4 – одна из удвоенных бедренных вен; 5 – тромб в просвете одной из удвоенных бедренных вен; 6 – подколенная вена [64].
Опорными жалобами, заставляющими внести ТЭЛА в дифференциально-диагностический ряд, являются одышка, болевой синдром (боли в грудной клетке различного характера, а порой – абдоминальные), частое сердцебиение. Часто отмечается падение артериального давления (возможно, с потерей сознания). К другим симптомам относятся кашель, кровохарканье, субфебрилитет, симптомы нарушения мозгового кровоснабжения и полиорганной недостаточности.
К сожалению, симптомы ТЭЛА не обладают ни высокой чувствительностью, ни специфичностью. В таблице 15 приведена распространённость наиболее характерных симптомов и данных физикального обследования у больных, поступивших с подозрением на ТЭЛА, в зависимости от заключительного диагноза. Очевидно, что ТЭЛА не является единственной возможной причиной перечисленных симптомов, и ни один симптом не встречается при ТЭЛА в 100% случаев. Таблица 15. Частота симптомов и признаков у больных, поступивших с подозрением на ТЭЛА, в соответствии с заключительным диагнозом [41].
| |||||||||||||||||||||||||||||||||||||||||||||