тесты. Хромосомные болезни. Аутосомные синдромы
 Скачать 1.17 Mb. Скачать 1.17 Mb.
|

                   Хромосомные болезни.Аутосомные синдромы Хромосомные болезни.Аутосомные синдромы1.Высокий лоб,микроцефалия,микрогнатия,маленький рот с опушенными уголками рта,крупные оттопыренные уши,задержкапсихомоторного развития.Какому синдрому характерны вышеприведенные признаки А.Патау Б.шерешевского-Тернера В.Эдвардса Г.Вольфа-Хиршхорна Д.Орбели 2.В медико-генетический центр обратились родители с подозрением на хромосомную болезнь ребенка.Врач установил диагноз:транслокауионная форма синдрома Дауна.Какой метод был использован для постановки диагноза? А.биохимический Б.генеологический В.молеклярно-цитогенетический Г.популяционно-статистический Д.близнецовый 3.В медико-генетическую консультацию обратилась беременная женщина,которая работала на вредном производстве и имела основания для волнения по поводу рождения ненормального ребенка.После проведения амиоцентеза встал вопрос о прерывании беременности.Врачи обьяснили женщине,что ее будущий ребенок не будет жизниспособным и будет иметь пороки в строении сердца,почек,пищеварительной системы.Расщепление мягкого и твердого неба,недоразвитие или отсутствие глаз.Определите кариотип А.47,ХУ,13+ Б.47,ХУ,18+ В.47ХХХ Г.47ХХУ Д.47,ХХ,21+ 4.При обследовании мальчика 1 месяца жизни,обнаружены долихоцефалия,еформированные ушные раковины,флексорное положение пальцев рук,врожденный порок сердца. Для какого из пречисленных синдромов характерны выявленные симптомы ? А.трисомия 13 Б.трисомия 18 В.трисомия 21 Г.синдром кошачьего крика Д.синдром орбели 5.У здоровых родителей родился ребенок с синдромом кошачьего крика.Определите к какому типу мутаций относится это нарушение. А.геномные Б.гоносомные В.генные Г.хромосомные Д.аутосомные 6.Какие из перечисленных заболеваний связаны с нарушением число хромосом: А.синдром Вольф-Хиршхорна Б.гемофилия В.синдром Орбели Г.дальтонизм Д.синдром Дауна 7.У 6-летнего ребенка обнаружен синдром Дауна.Но хромосомный анализ показал,что не все клетки имеют аномальный кариотип.Что из перечисленного наблюдается в данном случае? А.эпистаз Б.мозаицизм В.неполная пенетрантность Г.неполное доминирование Д.вариабальное экспрессивность 8.При изучении кариотипа установлена его формула 47,ХХ,18+.Определите,для какого синдрома характерно указанное изменение кариотипа: А.Эдвардса Б.Марфана В.кошачьи крик Г.Патау Д.Клайнфельтера 9.В медико-генетическую консультацую обратилась родителей новорожденного,у которого подозревают:синдромом Патау.Какой из перечисленных методов эффективен для определения диагноза? А.биохимический Б.генеалогический В.цитогенетический Г.определение Х-полового хроматина Д.близнецовый 10.Определите формулу кариотипа при мозаичной форме синдрома Дауна А.47,ХХ(+18)/46ХХ Б.47,ХХ(+18)/46ХУ В.47,ХУ(+13)/46,ХУ Г.47,ХХ(+21)/46,ХХ Д.45,Х0/46,ХХ Синдром Дауна – 47, XY + 21 или 47, XX + 21(трисомия по 21-й паре хромосом). Частота – 15 случаев на 10000 новорожденных обоих полов. Укороченные конечности, маленькая головка, плоское лицо, широкая переносица, монголоидный разрез глаз, большой язык, не помещающийся во рту. Аномалии строения внутренних органов. Резко выраженная умственная отсталость. Женщины иногда могут иметь детей, мужчины – никогда. 31% больных умирает до 1 года, причем от обычных простых заболеваний, так как снижен иммунитет. Вообще живут недолго. Иногда синдром Дауна обусловлен не трисомией, а транслокацией (реципрокной) между 13 – 15 и 21 хромосомами; в этом случае хромосом 46. Синдром Эдвардса – 47, ХХ + 18 или 47,XY + 18 (трисомия по 18-й паре хромосом). Частота – 1 случай на 7000 новорожденных обоего пола. Фенотип: множественные аномалии – деформация черепа, "птичий профиль" лица, короткие глазные щели, микрофтальм, низко расположенные и деформированные ушные раковины, короткая шея и грудина, врожденный вывих бедра, синдактилия, врожденные пороки сердца и крупных сосудов. Живут недолго: 30% умирает на первом и 50% – на втором месяце жизни Синдром Патау – 47, XY + 13 или 47, ХХ + 13(трисомия по 13-й паре хромосом). Частота – 1 случай на 6000 новорожденных обоего пола. Множественные уродства (микрофтальм, расщелина губы и неба, череп неправильной конфигурации, скошенный узкий лоб, плоский и широкий нос, запавшее переносье, низко расположенные уши, полидактилия, пороки развития внутренних органов). Живут несколько дней или недель. Синдром Лежьена – 46, ХХ, 5р- или 46,XY, 5р- (делеция короткого плеча ( р ) 5-й хромосомы). Частота – 1 случай на 50000 новорожденных обоего пола. Его еще называют «синдром кошачьего крика». Характеризуются многочисленными пороками развития черепа, строения гортани (в связи с чем при рождении вместо плача издают звук, похожий на кошачье мяуканье), конечностей, сердца, почек, глаз, тяжелой формой слабоумия. Живут такие больные недолго. Моногенные болезни с аутосомно-рецессивным типом наследования 1.На основании клинических данных врач заподозрил у новорожденного ребенка наследственное заболевание – галактоземию.Какой метод может подтвердить его предложение? А.цитогенетический Б.молекулярно-цитогенетический В.биохимический Г.клинико-генеалогический Д.определение Х-полового хроматина 2.Ребенок имеет отставание в росте и низкую массу тела,несмотря на нормальное потребление пищи,накопление густой липкой слизи,частые инфекции грудной клетки,кашель и одышку.Генетическое тестирование показывает мутацию обоих аллелей хромосомы.Диагноз ребенка А.кистозный фиброз Б.болезнь Реклинхаузена В.семейная гиперхолестеримию Г.фенилкетонурию Д.галактоземию 3.Известно,что альбинизм обусловлен аутосомно-рецессивной мутацией.Первый ребенок пары с нормальной пигментацией кожи был альбиносом.Какова вероятность того,что их второй ребонок также будет альбиносом? А.100% Б.75% В.0% Г.50% Д.25% 4.Одна из форм цистинурии наследуется как аутосомно-рецессивный признак,у гетеризигот наблюдается лишь повышенное содержание цистина в моче,а у гомозигот –образование цистиновых камней в почках.Определить возможные формы и вероятность проявления цистинурии у детей в семье,где оба родители имеют повышенное содержание цистина в моче А.Аа-100% Б.аа-100% В.Аа-50% аа-50% Г.АА-25% Аа-50% аа-25% Д.АА-25% Аа-25% аа-25% 5.Организмы,в генотипе которых представлены два разных мутантных аллеля одного гена называют А.компаунд Б.гомозигота В.гемизигот Г.гетерозигота Д.гетерокарион 6.Вероятность рождения больного ребенка в семье,в котрой оба родителя являются гомозиготами по генуфенилкутенории,составляет А.25% Б.75% В.100% Г.50% Д.0% 7.Известно,что нейросенсорная тугоухость обусловлена аутосомно-рецессивной мутацией.Известны семьи,где оба родителя больны,однако все дети здоровы.Это возможно обьяснить А.генетической гетерогенностью Б.полной пенетрантностью В.неполной пенетрантностью Г.вариебельной экспрессивностью Д.клиническим полиморфизмом 8.Вероятность рождения больного ребенка в семье в которой мать больна фенилкетонурией,а отец гомозиготен по нориальному аллелю,составляет А.100% Б.75% В.25% Г.0% Д.50% 9.Определить расщепление по генотипам среди детей,если один из родителей здоров,а у другого серповидклеточность А. АА-25% АS-50% SS-25% Б.SS-100% В.АА-50% SS-50% Г.АS-100% Д.АА-50% АS-50% 10.Наиболее ярким примером точечной мутации, проявляющейся в наследственной патологии,является А.синдром Дауна Б.серповидно-клеточная анемия В.врожденная талассемия Г,гемофилия А Д. Злокачественный опухоль Моногенные заболевания обусловлены мутациями или отсутствием отдельного гена. Мутации могут захватывать один или оба аллеля. Клинические проявления возникают в результате отсутствия генетической информации или реализации дефектной. Широкий круг моногенных болезней образуют наследственные нарушения обмена веществ, возникновение которых связано с мутацией генов, контролирующий синтез ферментов и обусловливающих их дефицит или дефект строения — ферментопатии. Моногенные болезни по типу наследования могут быть: - аутосомно-доминантными, - аутосомно-рецессивными, - сцепленными с полом; По фенотипическому проявлению могут быть: - ферментопатиями (болезнями обмена веществ, в т.ч. болезнями, обусловленными нарушением репарации ДНК), - болезнями, обусловленными молекулярной патологией структурных белков, - иммунопатологией, в т.ч. болезнями, вызванными нарушениями в системе комплемента; нарушениями синтеза транспортных белков (в т.ч. белков крови) и пептидных гормонов; патологией свертывающей системы крови, - дефектами механизма переноса веществ через клеточные мембраны. Моногенные болезни наследуются в полном соответствии с законами Менделя. Большинство известных наследственных болезней обусловлено мутациями структурных генов, возможность этиологической роли мутаций генов-регуляторов при некоторых заболеваниях до сих пор доказана лишь косвенно. Чаще всего аутосомно-рецессивные заболевания проявляют себя при браке типа Аа х Аа (оба родителя фенотипически здоровы, но являются носителями му тантного гена) в варианте образования го мозиготного организма (25%) по дефектному гену: 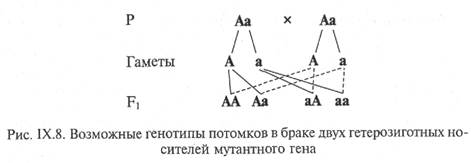 По аутосомно-рецессивному типу наследуется абсолютное большинство ферментопатий (наследственных заболеваний обмена ве ществ). Наиболее частыми и значимыми в клини ческом отношении являются такие болезни с аутосомно-рецессивным типом наследования, как муковисцидоз (кистофиброз поджелудочной железы), фенилкетонурия, адреногенитальный синдром, многие формы нарушения слуха или зрения, болезни накопления. Муковисцидоз(кистофиброз поджелудочной железы). Ген муковисцидоза, контролирующий синтез белка (трансмембранного регулятора проводимости) расположен на 7-й хромосоме. Заболевание обусловлено генерализованным по ражением экзокринных желез. При отсутствии синтеза трансмембранного регулятора (первичного продукта гена) нару шается транспорт хлоридов в эпителиальных клетках, что приво дит к избыточному выведению хлоридов, следствием чего являет ся гиперсекреция густой слизи в клетках поджелудочной железы, бронхов, слизистой оболочке желудочно-кишечного тракта. Вы водные протоки поджелудочной железы закупориваются, слизь не выводится, образуются кисты. Ферменты поджелудочной железы не поступают в просвет кишечника. Гиперпродукция слизи в брон хиальном дереве ведет к закупорке мелких бронхов и последующе му присоединению инфекции. Подобные процессы развиваются в придаточных пазухах носа и в канальцах семенников. В потовой жидкости повышена концентрация ионов натрия и хлора, что и является основным диагностическим лабораторным тестом. Интеллектуальное развитие у детей не страдает. Прогноз жизни при всех формах заболевания неблагоприятный. В настоящее время пациенты со смешанными формами редко живут более 20 лет. Частота его среди новорожденных в европейской популяции составляет 1:2500. Среди заболеваний с аутосомно-рецессивным типом наследования встречаются те, которые поражают различные системы органов и связаны с наличием комбинирован ных дефектов. Например, при синдроме Барде-Бидля наблюдается сочетанное на рушение умственного развития и зрения.Признаками синдрома являются: ожирение, гипогенитализм (нередко и гипогонидизм), умственная отста лость, пигментная дегенерация сет чатки, приводящая к потере зрения, и полидикталия со стороны 5 пальца. Из других нарушений зрения описаны ката ракта, атрофия зрительных нервов, нистагм. Уже на первом году жизни развивается ожирение, которое с возрастом прогрессирует. Характерным для синдрома является разнообразная патология по чек. Вероятность проявления таких заболеваний кратно возрастает при кровнородственных браках. В целом, в характеристике аутосомно-рецессивных заболеваний присутствуют следующие составляющие: 1) родители больного ребенка фенотипически здоровы, но являются носителями аномального гена в рецессивном состоянии; 2) мальчики и девочки заболевают одинаково часто; 3) риск рождения ребенка с аутосомно-рецессивным заболеванием составляет 25%; 4) отмечается «горизонтальное» распределение больных в ро дословной, т. е. пациенты чаще встречаются в рамках одного сибства; 5) наблюдается увеличение частоты больных детей в группах родителей, связанных родством, причем чем реже аутосомно-рецессивное заболевание встречается в популяции, тем чаще боль ные происходят из кровнородственных браков. Моногенные болезни , сцепленные с полом 1.Анализ родословной показал след.:у больного отца болеют только дочери ,а все сыновья и их дети здоровы.Определите тип наследования заболевания: Х-сцепленный доминантный 2.Причиной этой болезни является мутацией в коротком плече Х-хромосомы (Хр21) Дюшена 3.Передача признака имеет вертикальный характер наследования.Дочери никогда не наследуют признак от отца. Дальтонизм 4.Признак передается от отца всем сыновьям.Дочери никогда не наследуют от отца.Определите тип наследования признака Голандрический 5.Женщины передают патологический признак дочерям и сыновьям с вероятностью 50%,мужчины-только только дочерям с вероятностью 100%.У мальчиков болезнь протекает тяжелее,чем у девочек.Определите тип наследования признака Х-сцепленный доминантный 6.Если у женщины обнаружен в крови высокий уровень креатинфосфаткиназы,каков риск рождения мальчика с мышечной дистрофией Дюшенна 50% 7.Мужчины могут наследовать этот ген только от матери.Женщины при этом типе наследования передаютпатологический признак сыновьям,определите тип насл-я признака Х-сцепленное рецессивный 8.Первые признаки заболевания проявляются в 10-15 летнем возрасте,иногда раньше.Начальные симптомы-мышечная слабость,патологическая мышечная утомляемость при физ.нагрузке,псевдогипертрофия икроножных мышц.Заб-е иедленно прогрессириует.Темп распр-я атрофии невысок,и больные длительное время сохраняют работоспособность. Беккера 9.Определить генотипы женщины и ее сына,больного миодистрофией Дюшенна хАха хаУ 10.Болезнь у ребенка начинает проявляться не сразу после рожд-я,а только в возрвсте1,5-5 лет,признаки можно считать такие проявления ,как постоянные падения при ходьбе.Если ребенку нужно подняться по лестнице,то он старается этого избегать.Это характерно для синдрома : Дюшенна Сцепленные с полом: 1)х-сцепленные (А). Ген расположен в Х хромосоме, наследоваться может в каждом поколении. 2) х-сцепленные (а). Заболевания с рецессивным типом наследования проявляются у лиц мужского пола в гемизиготном состоянии. Гемофилия, дальтонизм (нарушение цветовосприятия не различимы красный и зеленый). 3)у-сцепленный тип. Ген перепонок между пальцами, ген оволоснения. В У – хромосоме находятся главные гены формирующие мужские половые признаки. Предтесты по «Медицинской генетике» 2017-2018 уч. Год |