Клiнiчна_фармакологiя_навчал_нии_посiбник. Навчальний посібник до практичних занять для студентів медичних факультетів вищих медичних навчальних закладів
 Скачать 466.6 Kb. Скачать 466.6 Kb.
|

І. Теоретичні питанняВизначення предмету і завдань клінічної фармакології. Основні принципи фармакокінетики. Основні фармакокінетичні параметри: біодоступність, кліренс, об’єм розподілу, період полужиття. Основні фармакокінетичні процеси: всмоктування, розподіл, біотрансформація, екскреція лікарських засобів. Основні принципи фармакодинаміки. Механізм дії лікарських речовин, їх фармакологічні ефекти та зміни стану функцій організму у відповідь на вплив лікарських препаратів. ІІ. Теоретичний матеріал2.1. Предмет і завдання клінічної фармакології Клінічна фармакологія як предмет розглядає проведення максимально раціональної лікарської терапії у конкретного хворого, методологією вибору методу лікування з врахуванням індивідуальних особливостей організму, перебігу та форми захворювання, наявності супутньої патології, визначає найбільш ефективні та безпечні лікарські засоби, а також їх комбінацій, на основі даних доказової медицини. Основні завдання клінічної фармакології включають проведення клінічних випробувань фармакологічних засобів та розробку методів ефективного та безпечного застосування ліків. Основними розділами клінічної фармакології є фармакодинаміка і фармакокінетика. Серед лікарів добре відомо, що при дія ліків може сильно розрізнятися серед різних індивідуумів. Ліки впливають на специфічні таргетні молекули, що призводить до основних та побічних фармакологічних ефектів. Процеси між введенням препарату і розвитком цих ефектів можна розділити на два важливих компонента, кожен з яких відповідає за варіабельність дії ліків. Сукупність ефектів фармакологічного засобу і механізм його дії відносяться до фармакодинамічних процесів. Ці властивості ліків визначають групу, до якої відноситься лікарський засіб і часто мають важливу роль у визначенні, чи є дана група адекватною терапією конкретного симптому чи захворювання. Фармакокінетичні процеси визначають вплив організму на лікарський засіб та включають вивільнення ЛР із з лікарської форми всмоктування, розподіл, біотрансформацію та екскрецію. Крім того, клінічна фармакологія вивчає побічні дії, особливості дії ліків у залежності від різноманітних умов (вік, захворювання, вагітність), взаємодію ліків при їх сумісному застосуванні, вплив їжі та фармакологічний ефект та інше. Відносно новий розділ клінічної фармакології – фармакогенетика, предметом якої є визначення генетичних основ реакції організму на лікарський засіб. 2.2. Основні принципи фармакокінетики 2.2.1. Основні фармакокінетичні параметри Концентрація лікарської речовини (С) – це її кількість у певному об’ємі крові в конкретний момент часу після введення в організм. Об’єм розподілу препарату (Vd – volume of distribution) – характеризує ступінь захоплення лікарської речовини (ЛР) тканинами з плазми крові і виражається формулою Vd=D/Co – це умовний об’єм рідини, потрібний для рівномірного розподілу введеної дози ЛР до концентрації, що визначається у крові в момент дослідження. Розрізняють також поняття питомий об’єм розподілу – це відношення об’єму розподілу до маси тіла, виражається у л/кг. Об’єм розподілу ЛР певною мірою визначає ступінь проникнення її з крові й позаклітинної рідини у тканини, а також створення її депо в органах. Площа під фармакокінетичною кривою "концентрація - час" (AUC – area under the curve) - площа фігури, обмежена фармакологічною кривою і осями координат (АUC = Co/Kel). Площа під кривою залежить від об’єму розподілу і загального кліренсу лікарського засобу. Біодоступність (F) визначають відносною кількістю ЛР, що надходить до системного кровообігу і взаємодіє з тканинними рецепторами. Біодоступність лікарського засобу при позасудинному введенні визначають як співвідношення до його лікарської форми для внутрішньосудинного введення. Якщо досліджують лікарські форми для позасудинного і внутрішньовенного введення у рівних дозах, застосовують формулу:  Біоеквівалентність (порівняльна біодоступність) – це співвідношення кількості ЛР, що надходить у кров при введенні її в різних лікарських формах (або лікарських препаратів різних фірм). Якщо лікарські препарати демонструють схожу біодоступність, вони розцінюються як біоеквівалентні. Загальний кліренс препарату (Cl) – це умовний об’єм крові чи її плазми, що очищається від ЛР за одиницю часу. Він характеризує швидкість елімінації лікарського препарату з організму. Виражають кліренс у літрах за годину чи мілілітрах за 1 хвилину і розраховують за формулою: 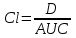 де Сl – загальний кліренс; D – доза уведеного препарату; AUC – площа під фармакокінетичною кривою. Елімінація ліків відбувається у нирках, печінці та інших органах (легені, потові залози, молочні залози тощо). Тому виділяють нирковий, печінковий та інші види кліренсів, що в сумі складають загальний кліренс: 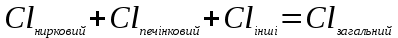 Кліренс у різних органах залежить від швидкості кровообігу у органі (Q) та коефіцієнта екстракції препарату органом (ER): 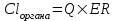 Коефіцієнт екстракції залежить від кількості препарату, що поступає в орган з артеріальною кров’ю (СА) і кількості препарату, що виводиться з органу з венозною кров’ю (СV): 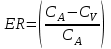 Період напіввиведення (Т1/2 абоT0,5) – це фармакокінетичний показник часу, протягом якого кількість ЛР в камері або його концентрація в крові зменшується на 50 %. 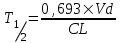 , ,де Т1/2 – період напіввиведення; 0,693 – коефіцієнт, що є логарифмом від 2; Vd - об’єм розподілу; Сl - загальний кліренс. Константа елімінації (Кel) – відсоток зменшення концентрації ЛР в крові за одиницю часу. Чим більша Кel, тим швидше ЛР видаляється з крові. Константа елімінації залежить від періоду напіввиведення: 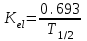 Період напівабсорбції (Т1/2а) - час, необхідний для всмоктування половини дози лікарського засобу з місця введення в системний кровоток; пропорційний швидкості абсорбції (Т1/2а = 0,693/Ка). Константа абсорбції (Ка) – характеризує швидкість всмоктування ЛР в організмі людини чи тварини. Константа абсорбції залежить від періоду напіввиведення: 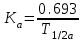 Удавана початкова концентрація (С0) – концентрація препарату, що була б у плазмі крові при внутрішньовенному шляху введення і миттєвому розподілі по органах і тканинам. Рівноважна концентрація (Сss) – концентрація препарату, що встановлюється в плазмі крові при надходженні препарату в організм із постійною швидкістю. При переривчастому введенні препарату через однакові проміжки часу в однакових дозах виділяють максимальну (Сmax) і мінімальну (Сmin) рівноважні концентрації. 2.2.2. Всмоктування лікарських речовин Всмоктування (абсорбція) лікарських речовин – проникнення ліків через біологічні мембрани в судинне русло. Швидкість вивільнення з лікарської форми з різних ЛР неоднакова. Процес вивільнення лімітує швидкість всмоктування в тих випадках, коли ліки даються в твердій формі. Наприклад, вивільнення ЛР з таблетки включає як процес розпаду, так і процес розчинення. На швидкість розчинення впливають певні характеристики складу лікарської форми. При цьому важливими є розмір і форма частинок, форма кристалів і такі добавки, як забарвлюючі, ковзні, розпушуючі і суспендуючі речовини, а також виробничі змінні: тиск пресування, вміст вологи у таблетках тощо. Природно, що і ступінь всмоктування ліків неоднаковий, оскільки на нього впливають такі фактори, як моторика шлунково-кишкового тракту (ШКТ) і швидкість проходження. При ентеральному введенні всмоктування відбувається головним чином в тонкому кишківнику. При всмоктуванні відбувається як пасивний, так і активний енергозалежний транспорт. Для перенесення речовин в ШКТ особливе значення мають велика площа поверхні кишківника і вплив постійного кровотоку в слизовій оболонці на градієнти концентрації між просвітом кишківника і кров’ю. Шляхом дифузії і осмосу через слизову оболонку кишківника переносяться, зокрема, вода, С1¯, а також такі речовини, як аскорбінова кислота, піридоксин і рибофлавін. Оскільки клітинні мембрани містять велику кількість ліпідів, для дифузії через мембрану речовини повинні бути в деякій мірі жиророзчинними. Згідно теорії неіонної дифузії, вказаним шляхом переносяться головним чином недисоційовані солі слабких кислот або слабких основ. Це необхідно враховувати при призначенні ліків, велика частина яких всмоктується шляхом дифузії. Для перенесення якоїсь речовини відповідно до рівняння Гендерсона-Гассельбаха особливе значення має рКа цієї речовини і рН в просвіті кишківника: 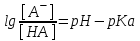 , , 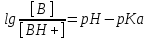 , де , де[А¯], [ВН+] – молярні концентрації іонізованих, [НА], [В] – неіонізованих форм кислоти НА і основи В; рН – кислотно-основний показник середовища; рКа – логарифм константи дисоціації сполуки, чисельно рівний значенню рН, при якому аналізована сполука дисоціює наполовину. З рівняння видно, що із збільшенням значення рН середовища дисоціація кислот збільшується, а основ – зменшується. Таким чином, фактори, що впливають на процеси всмоктування ЛР, різноманітні: розчинність речовини в ліпідах, ступінь іонізації молекули (чим менше іонізована молекула, тим краще вона всмоктується), перистальтика кишківника, характер і кількість харчової маси, особливості реґіонарного кровотоку, стан сполучної тканини, агрегатний стан речовин, поєднання лікарських засобів. На всмоктування можуть впливати ступінь наповнення шлунку, здатність ЛР до комплексо-, хелато- і іоноутворення, а також об’єм, склад і в’язкість секрету, ступінь взаємодії з активним транспортом, проникність слизової оболонки травного тракту, ушкоджувальна дія препарату і харчових продуктів на слизову оболонку, вплив на мікрофлору, що бере участь в метаболізмі препарату. Процес всмоктування залежить також перистальтики, місцевого кровообігу, наявності ферментів тощо. Як правило, вказані фактори взаємозв’язані і обумовлені індивідуальними і віковими особливостями хворого, специфікою перебігу патологічного процесу. Ліки, що всмокталися в порожнині рота або в прямій кишці, проходять через примикаючі капілярні мережі безпосередньо у велике коло кровообігу, що дозволяє усунути пресистемний метаболізм. При сублінгвальному введенні препарат проникає в системний кровообіг через вени голови, які впадають в яремну вену. Отже, такі ліки (наприклад, нітрогліцерин) не метаболізуються під дією ферментів печінки або кишківника до їх надходження в загальний кровообіг. Лікарський препарат, введений перорально, піддаватиметься інтенсивному метаболізму,а при всмоктуванні в порожнині рота або в прямій кишці в повнішому об’ємі поступає в системний кровообіг. Інші відділи ШКТ відрізняються один від одного величиною рН секрету, властивостями поверхневого епітелію, ферментами і в результаті – здатністюабсорбувати різні ЛР. Шлунковий сік людини має в нормі рН 1-3, вміст дванадцятипалої кишки досягає рН 6-8. а вміст тонкої і товстої кишок рН близько 8. Тому препарати-кислоти краще всмоктуватимуться у шлунку, а ліки-основи – в кишківнику. Невеликі нейтральні молекули, наприклад спирту і води, добре всмоктуються у шлунку. Кисле середовище шлунку, окрім впливу на ступінь іонізації ЛР, може викликати їх хімічне руйнування (наприклад, бензилпеніцилін). При внутрішньом’язовому введенні водних розчинів гідрофільних препаратів спостерігається їх швидке всмоктування в кров. З олійних розчинів ліпофільні препарати всмоктуються повільніше, утворюючи в м’язах депо. Швидше всмоктування спостерігається при введенні препарату в м’язи стегна, ніж при ін’єкції в м’язи сідниці. Всмоктування через шкіру використовується для створення не тільки місцевого, але і системного ефектів. У разі підшкірної ін’єкції ліки, розчиняючись в тканинній рідині, всмоктуються в капіляри і лімфатичні судини дерми. Шляхом інгаляції можуть вводитися ЛР у вигляді аерозолів, газів і порошків. У легенях всмоктуються газоподібні і леткі речовини, що використовуються для наркозу (ефір, хлороформ, азоту закис, фторотан тощо). Основним показником всмоктування є біодоступність – відносна кількість ЛР, яка досягає системного кровотоку. Окрім властивостей самої речовини, на біологічну доступність можуть впливати технологія виготовлення лікарської форми, взаємодія з їжею і інші умови. Біодоступність лікарського засобу після внутрішньовенного введення завжди дорівнює 100 %. Тому на практиці біодоступність при однакових дозах лікарського препарату визначають за формулою: 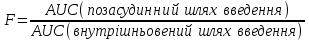 де AUC – площа під кінетичною кривою «концентрація–час» (area under the curve). Також виділяють порівняльну біодоступність (біоеквівалентність) – показник порівняння біодоступностей двох лікарських форм одного лікарського засобу. Наприклад, різні лікарські форми: таблетки, капсули, розчин однієї і тієї ж сполуки можуть відрізнятися за біодоступністю. 2.2.3. Розподіл лікарських речовин Розподіл лікарських речовин – рух речовини від місця всмоктування до міста дії і, далі, до міста елімінації. Ступінь тканинної проникності для лікарських засобів залежить від ряду чинників: величини концентрації препарату в крові, ступені їх зв’язування з білками плазми, міжклітинних просторів і цитоплазми клітин-мішеней, швидкості проникнення через різні біомембрани, швидкості кровоплину в тканинах, наявності тих або інших патологій, що змінюють ці показники (рівня або структури сироваткових і тканинних білків, з якими зв’язуються дані ліки, мембранної проникності, кровоплину в місці інфекції тощо). 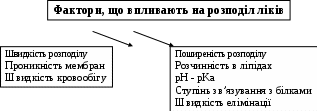 Фізико-хімічні властивості препарату (молекулярна маса, рівень іонізації і полярності, розчинність у воді і ліпідах) впливають на його проходження через мембрани, а отже, і на розподіл. На величину розподілу впливають і фізіологічні чинники – вік, стать, загальна кількість жиру в організмі. Крім того, розподілзмінюється при деяких патологічних станах, особливо при захворюваннях печінки, нирок, серцево-судинної системи і ін. Основним показником розподілу є об’єм розподілу (Vd – volume of distribution) – об’єм рідини, у якому розчиняється лікарський засіб з концентрацією, що дорівнює його концентрації у плазмі крові. При судинному введенні величина Vd не може бути менше об’єму кровіі при значеннях Vdблизько 5 % від об’єму організму доречно припустити, що розподіл речовини обмежується об’ємом кров’яного русла. Величини Vd, у межах 15-30 %, свідчать про об’ємний розподіл препарату в позаклітинній рідині, а Vd = 50-60 % – у всій водній фазі організму.Якщож величина Vdперевищуєреальний об’єм організму то слід припустити, що відбувається кумуляція речовини поза судинним руслом. У клінічній практиці Vdвикористовуєтьсядля розрахунку навантажувальної дози, потрібної для досягнення необхідної концентрації препарату в крові: D = Vd ·C де D – доза; Vd– об’єм розподілу; C – необхідна концентрація. Розподіл ліків тісно пов’язаний з проникненням ліків через біологічні бар’єри організму: гематоенцефалічний, плацентарний, гематоофтальмічний, тестикулярний. Гематоенцефалічний бар’єр утворюють безперервний ендотелій капілярів, клітини якого сполучені обширними щільними контактами, які,майже повністю перекривають щілини і дуже щільна базальна мембрана, що оточує капіляри. Проникність мозкових оболонок підвищена в ранньому дитячому і старечому віці, при гострому запальному процесі, знижена – при утворенні капсули фібрину навколо абсцесу мозку. Відзначають високі концентрації як в тканині мозку, так і в гнійному вмісті абсцесів високоліпофільних препаратів, що слабо зв’язуються сироватковими білками, а також ліків з низькою молекулярною масою (левоміцетин, метронідазол, кліндаміцин, фузидієва кислота). У нормі ліки порівняно погано проникають в глибокі шари ока. Навіть при місцевому застосуванні, коли концентрація в тканинах ока вища, ніж при парентеральному введенні, визначити препарат, наприклад, в склоподібному тілі не завжди вдається. У проникненні препаратів через плевру відіграють роль різні чинники: зв’язування з білками крові, молекулярна маса ліків, їх ліпофільність, зв’язування з рецепторами клітинних мембран тощо. Як відомо, в нормі кров матері і плоду не змішується, між ними існує плацентарний бар’єр. Механізми трансплацентарного перенесення аналогічні механізмам проникнення через інші бар’єри – пасивний транспорт, полегшена дифузія, активний транспорт, ендоцитоз; деякі низькомолекулярні речовини можуть проходити через водні пори в плаценті. Гідрофільні і іонізовані молекули ліків дуже слабо проникають через плаценту. Багато нейтральних речовин, слабкі кислоти або основи більшою чи меншою мірою проходять через плаценту, викликаючи у плоду ембріотоксичну або специфічну фармакологічну дію. Білки плазми крові мають специфічну структуру і своїми активними групами можуть зв’язуватися з ліками. Швидкість, ступінь і міцність зв’язування залежать від конформації і комплементарності цих центрів і характер хімічних зв’язків, що виникають при взаємодії. Тільки незв’язані речовини можуть дифундувати в тканини, оскільки комплекс білок-ліки не здатний пройти через мембрану клітини. Ступінь зв’язування білків з ліками залежить від наступних чинників: кількості різних типів, що зв’язують макромолекули; концентрації макромолекул кожного типу; здатності зв’язувати або «спорідненості» активного центру білка і лікарського препарату; наявності конкуруючих хімічних сполук екзогенного характеру; фізико-хімічного стану крові: рН, температури, іонного складу, в’язкості, осмотичного тиску тощо. Ряд тканинних структур здатні активно зв’язувати певні хімічні речовини. Наприклад, тканина щитовидної залози накопичує сполуки міді, кісткова тканина – тетрациклін і т.д. Зменшення кількості білків плазми, що зв’язують ліки на 10-15 % спостерігається при старінні. Це забезпечує збільшення концентрації в плазмі лікарських препаратів при стандартній дозі і розвитку побічних ефектів. Зв’язування лікарських засобів з білками крові порушується при деяких захворюваннях (опіки, нефротичний синдром, хронічні захворювання печінки, множинна мієлома). 2.2.4. Біотрансформація ліків Біотрансформація (метаболізму) ліків включає біохімічні процеси перетворення ліків зі зміною їх фармакологічних властивостей і утворенням метаболітів, які можуть виводитися з організму. В результаті біотрансформації утворюються терапевтично активні, індиферентні чи токсичні продукти. При цьому речовини отримують більшу полярність і, як наслідок, вищу гідрофільність. Розрізняють два типи реакцій метаболізму лікарських препаратів в організмі: несинтетичні (реакції I фази) і синтетичні (реакції II фази). Усі несинтетичні реакції метаболізму лікарських препаратів можна розділити на дві групи: реакції, що каталізуються ферментами ендоплазматичного ретикулуму (мікросомні), і реакції, що каталізуються ферментами, локалізованими в інших місцях (немікросомні). Несинтетичні реакції (I фаза) обумовлюють специфічну перебудову в молекулі субстрату з утворенням функціональних груп з активним атомом водню – оксигруп, первинних чи вторинних аміногруп, карбоксигруп тощо. У синтетичних реакціях (II фаза) по функціональних угрупованнях, що утворилися, проходить кон’югація з високополярними кислотними залишками, наприклад, із залишками глюкуронової, сірчаної, деяких амінокислот. Існує кілька типів окислювально-відновних ферментативних реакцій. Ці реакції каталізуються ферментами системи цитохрому Р450 (або CYP), флавінмонооксигенази та епоксидгідролази. Класифікація ферментів CYP-450 базується на особливостях структури: якщо співпадають 40-55 % амінокислот, ферменти відносять до однієї групи, якщо більше 55 % – до однієї підгрупи. Виділяють чотири родини ферментів CYP-450: • Група 1: CYP1A • Група 2: CYP2A, CYP2B, CYP2C, CYP2D, CYP2E • Група 3: CYP3A • Група 4: CYP4A З цієї класифікації зрозуміло, що родини CYP1, CYP3 і CYP4 мають по одній групі і CYP2 має п’ять груп. Нижче приведені найбільш важливі представники підгруп: • Підгрупа CYP1A: CYP1A1, CYP1A2 • Підгрупа CYP2A: CYP2A1, CYP2A5, CYP2A6 • Підгрупа CYP2B: CYP2B1, CYP2B2, CYP2B6 • Підгрупа CYP2C: CYP2C8, CYP2C9, CYP2C18, CYP2C19 • Підгрупа CYP2D: CYP2D6, CYP2E1 • Підгрупа CYP3A: CYP3A4, CYP3A5, CYP3A7 • Підгрупа CYP4A: CYP4A9, CYP4A11 CYP3A4 бере участь у біотрансформації більшості ліків, велика його кількість знаходиться за межами печінки. Підвищення метаболізму за рахунок CYP3A4 у шлунково-кишковому тракті може бути причиною зниження біодоступності багатьох ліків. Гідролази (Пептидази). Глікозид-гідролази каталізують гідролітичне розщеплення і називаються відповідно до типу зв’язку, що розривається. Ліази відщеплюють групи від молекули субстрату негідролітично. Вони також утворюють подвійні зв’язки чи приєднують групи по подвійних зв’язках. Вони можуть відщеплювати СО2, Н2о, NH3 і більш складні групи. Трансферази переносять групи атомів за допомогою специфічних переносників, що діють як коферменти. Вони відіграють роль у біохімічних перетвореннях і можуть переносити метильні, карбоксильні, аміно-, сульфо-, формільні чи фосфорильні групи. Основним органом, у якому відбувається метаболізм лікарських препаратів, є печінка. Менше значення мають нирки, м’язова тканина, стінка кишківника і легені. Лікарські засоби ще до досягнення системного кровообігу можуть бути метаболізовані в епітелії ШКТ чи в печінці. Даний процес, названий ефектом першого проходження, знижує біологічну активність ліків. Позаяк лікарські засоби, що призначаються всередину, до надходження в системну циркуляцію проходять через печінку, їх можна розділити на дві групи: перша – з високим печінковим кліренсом, друга – з низьким. Здатність печінки метаболізувати препарати першої групи залежить від швидкості їхньої доставки до печінки, тобто від печінкового кровотоку. Кінетика таких препаратів значно змінюється при захворюваннях, що порушують печінковий кровоток. Для другої групи лікарських препаратів печінковий кліренс залежить не від швидкості печінкового кровотоку, а від ємкості ферментативних систем печінки, що метаболізують дані препарати. На біотрансформацію лікарських засобів в організмі впливають багато чинників – вік, стать, зовнішнє середовище, характер харчування, захворювання тощо. Як відомо, у плода і немовляти в печінці порівняно мало паренхіматозних і значно більше ретикулоендотеліальних клітин, тому печінка дитини витягує менше речовини з кровоносного русла й у меншому ступені затримує її у своїх клітинах. У печінці плоду і немовляти відзначається менша активність окислювальних ферментів, ніж у дорослої людини. У зв’язку з цим у I фазі утвориться менше гідроксильованих метаболітів і з’являються вони з меншою швидкістю, що затримує інактивацію шляхом утворення парних ефірів. Активність глюкуронідазної системи, що приводить до утворення глюкуронідів, розвинута недостатньо не тільки у немовлят, але й у дітей до 12 років. Тому в дітей значно повільніше метаболізуються бутамід, амідопірин, діазепам, левоміцетин тощо. Разом з тим у них у печінці можуть утворюватися незвичайні метаболіти, яких у нормі не виявляються в дорослих. Сульфатів (ацетиламінофену і ін.) у немовлят утворюється достатньо, але в процесі постнатального життя процес глюкуронізації стає переважаючим. У процесі старіння організму людини відбуваються зміни кількості і розмірів частини клітинних елементів паренхіми, частково порушується кровоплин через печінку, знижуються її функції, у тому числі білокутворювальна й антитоксична. Зміна активності ферментних систем, що метаболізують ЛР, є однією з причин, що сповільнюють біотрансформацію ліків в організмі людей похилого і старечого віку й сприяють підвищенню їхньої концентрації в крові й у тканинах. Під час вагітності знижується метаболізм ЛР в організмі людини. Зниження активності оксидаз і глюкуронілтрансферази може бути обумовлено високим рівнем гормонів при вагітності, а саме: прегнандіолу, прогестерону і естрогенів, що є субстратами для зазначених ферментів і здатних гнітити метаболізм ендогенних речовин. Особливий практичний і теоретичний інтерес має з’ясування характеру змін біотрансформації ЛР при різних патологічних станах. При захворюваннях печінки, наприклад при цирозах, порушується не тільки функція печінкових клітин, але і печінковий кровоплин. Тому особливо змінюється фармакокінетика препаратів з високим печінковим кліренсом. До теперішнього часу встановлений ряд спадкових генетичних дефектів обміну, що призводять до атипічних реакцій на деякі лікарські препарати, вивченням яких займається наука фармакогенетика. Недостатність глюкозо-6-фосфатдегідрогенази (Г-6-ФД) відноситься до найпоширенійших фармакогенетичних дефектів. Носіями такого генетичного дефекту є не менш 200 млн. чоловік, найчастіше він зустрічається в жителів Середземноморського регіону. Сутність патологічного процесу при недостатності Г-6-ФД полягає в тому, що прийом деяких лікарських препаратів веде до масивного руйнування еритроцитів (тобто гемолітичних кризів). Тому при призначенні таких препаратів, як саліцилати, нітрофурани, сульфаніламіди, аскорбінова кислота, метиленовий синій, нітрати, левоміцетин, фенацетин, потрібна певна обережність. Недостатність ацетилтрансферази. Було виявлено, що переносимість тубазиду хворими неоднакова, у деяких хворих виникають важкі побічні явища: головний біль, блювота, біль за грудиною, поліневрит. Це явище має місце при дефекті ферменту, який інактивує ізоніазид – ацетилтрансферази, що необхідно враховувати при лікуванні туберкульозу. Зазначений фермент ацетилує також сульфаніламіди, новокаїнамід. Недостатність глюкуронілтрансферази. В основі спадкової негемолітичної жовтяниці лежить недостатність глюкуронілтрансферази – ферменту, що забезпечує утворення глюкуронідів білірубіну і багатьох лікарських засобів. Деякі ліки (стрептоміцин, хлорамфенікол, прогестерон) гальмують і без того різко знижену активність ферменту, у зв’язку з чим їхнє застосування таким хворим протипоказане. Біотрансформація деяких лікарських засобів (кортизон, хлормицетин) порушена: вони не перетворюються в глюкуроніди і кумулюють в організмі. Дози таких препаратів при даній патології повинні бути значно знижені. Інші спадкові порушення біотрансформації ліків включають недостатність редуктази метгемоглобіну, каталази, бутирилхолінестерази, оксидаз. 2.2.5. Екскреція ліків Розрізняють кілька шляхів виведення (екскреції) лікарських речовин і їхніх метаболітів з організму. До основних відносяться виведення з калом і сечею, менше значення має виведення з видихуваним повітрям, потом, слиною і слізною рідиною. Виведення нирками. Лікарські речовини виводяться з сечею шляхом клубочкової фільтрації і канальцевої секреції. Велике значення має також їхня реабсорбція в канальцях нирок. Кров, що потрапляє в нирки, фільтрується в клубочках. При цьому ЛР проникають через стінку капілярів у просвіт канальців. Фільтрується тільки та частина ЛР, що знаходиться у вільному стані. При проходженні через канальці частина ЛР реабсорбується і повертається в плазму крові. Багато ЛР активно секретуються з капілярів і перитубулярної рідини в просвіт канальців. При нирковій недостатності клубочкова фільтрація знижується, і виведення різних ЛР порушується, що приводить до збільшення їхньої концентрації в крові. Дозу препаратів, що виводяться із сечею, при прогресуванні уремії варто знизити. Слабкі кислоти швидше виводяться при лужній реакції сечі, а слабкі основи – при кислій. Виведення з жовчю. З печінки ЛР у вигляді метаболітів чи у незміненому вигляді надходять у жовч шляхом пасивного транспорту чи за допомогою активних транспортних систем. Надалі ЛР чи їхні метаболіти виводяться із організму з калом. Під впливом ферментів ШКТ чи бактерійної мікрофлори вони можуть перетворюватися в інші сполуки, що реабсорбуються і знову поступають в печінку, де вступають у новий цикл метаболічних перетворень. Подібний цикл називається ентерогепатичною циркуляцією. На виведення ЛР з жовчю впливають молекулярна маса сполуки, її хімічна природа, стан гепатоцитів і жовчовивідних шляхів, інтенсивність зв’язування ЛР з клітинами печінки. 2.3. Основні принципи фармакодинаміки Фармакодинаміка – розділ клінічної фармакології, який вивчає механізм дії і фармакологічний ефект ЛР. Основні механізми дії ліків включають: дія на специфічні рецептори (агоністи та антагоністи); вплив на активність ферментів (індукція і ігибування); вплив на мембрани клітин; пряма хімічна взаємодія ліків. 2.3.1. Види фізіологічних рецепторів Ефект більшості лікарських препаратів є результатом їх взаємодії з макромолекурярними компонентами клітинних мембран. Ця взаємодія викликає біохімічні та фізіологічні зміни, що характеризують ефект препарату. Термін рецептор застосовується до клітинної макромолекули, з якою препарат зв’язується для досягнення його ефекту. Протеїни відіграють найважливішу роль у формуванні рецепторів. Найбільш важливою групою рецепторів для ліків є протеїни, що фізіологічно працюють як рецептори ендогенних регуляторних лігандів (наприклад, рецептори гормонів, нейротрансмітерів). Багато ліків діють на такі рецептори і часто є високоселективними завдяки специфічності фізіологічних рецепторів. Регуляторна активність рецептора може проявлятися як наслідок прямої дії на клітинні мішені, ефекторні протеїни, або через проміжні клітинні сигнальні молекули (трансдуктори). Взаємодію рецептора, клітинної мішені та проміжних молекул розглядають як рецептор-ефекторну систему. Рецептори пов’язані з G-протеїном. Велике сімейство рецепторів для багатьох існуючих ліків (біогенні аміни, ейкозаноїди, пептидні гормони, опіоїди, амінокислоти) включає гетеротримерні регуляторні протеїни, що пов’язані з гуанінтрифосфатом (G-протеїни). G-протеїни є сигнальними трансдукторами, що передають інформацію від рецептора ефекторним протеїнам, таким як аденілатциклаза, фосфоліпаза С, фосфодіестерази, Са2+- та К+-іонні канали мембрани. Рецептори для ферментів. Велика група рецепторів з внутрішньою ферментною активністю включає протеїнкінази клітинної поверхні, що розповсюджують регуляторні сигнали через ефекторні протеїни на внутрішній поверхні клітинної мембрани. Фосфорилування протеїнів може змінювати біохімічну активність ефектора, або його взаємодію з іншими протеїнами. Більшість рецепторів, що є протеїнкіназами, фосфорилують тирозин в субстраті. Ця група включає рецептори до інсуліну, факторів росту. Деякі рецепторні протеїнкінази фосфорилують серин і треонін. Для рецепторів, що зв’язують передсердні натрійуретичні пептиди, гуанілін та урогуанілін, внутріклітинною структурою є гуанилілциклаза, а не протеїнкіназа. Гуанилілциклаза бере участь в секреції вторинного месенжера циклічного гуанозинмонофосфату (ГМФ), який активує циклічну ГМФ-залежну протеїнкіназу і активує декілька нуклеотідних фосфодіестераз. Іонні канали. Рецептори для деяких нейротрансмітерів формують селективні іонні канали. Ця група включає нікотинові холінергічні рецептори, рецептори ГАМК, рецептори для глутамату, аспартату та гліцину. Рецептори що регулюють транскрипцію. Рецептори для стероїдних та тироїдних гормонів, вітаміну D, ретиноїдів – це розчинні протеїни, що зв’язуються з ДНК та регулюють транскрипцію специфічних генів. Більшість рецепторів в структурі мають протеїни, їх агрегати і комплекси з нуклеїновими кислотами та низькомолекулярними сполуками. 2.3.2. Сигналізація за участю поверхневих рецепторів клітин і вторинних посередників Водорозчинні сигнальні молекули, зокрема всі відомі нейрорегулятори, пептидні гормони і багато інших ліків приєднуються до специфічних білкових рецепторів на поверхні клітин-мішеней. Поверхневі рецептори зв’язують сигнальну молекулу (ліганд), проявляючи велику спорідненість до неї, і ця позаклітинна подія породжує позаклітинний сигнал, що змінює поведінку клітини. Оскільки вказані рецептори є нерозчинними інтегральними мембранними білками і складають зазвичай менше 1% загальної маси білків плазматичної мембрани, їх важко виділити і вивчити. Число рецепторів конкретного ліганда може варіювати в межах від 500 до 100000 і більше на клітину; відразу після зв’язування ліганда рецептори можуть розташовуватися на плазматичній мембрані випадковим чином або скупчуватися в певних її ділянках. Багато білкових сигнальних молекул потрапляє всередину клітин-мішеней шляхом ендоцитозу, опосередкованого рецепторами. Але деякі сигнальні молекули можуть впливати на клітини, не проникаючи в них. Переважна більшість поверхневих рецепторів для гідрофільних сигнальних молекул, зв’язавши ліганд на зовнішній стороні мембрани, зазнає конформаційних змін. Такі зміни створюють внутрішньоклітинний сигнал, який змінює поведінку клітини-мішені. Внутрішньоклітинні сигнальні молекули часто називають вторинними посередниками, вважаючи «первинним посередником» позаклітинний сигнал. Відомо два загальні способи створення внутрішньоклітинного сигналу поверхневими рецепторами. Один з них полягає в активації або інактивації ферменту, пов’язаного з плазматичною мембраною. Цей механізм працює головним чином в електрично активних клітинах, наприклад в нейронах і м’язових волокнах. В деяких випадках вказаний фермент каталізує утворення розчинного внутрішньоклітинного медіатора, зміна концентрації якого служить сигналом. Другий спосіб дії рецепторів полягає в тому, що вони відкривають або закривають регульовані іонні канали плазматичної мембрани. 2.3.3. Адаптація клітин-мішеней Клітини-мішені, що зазнавали дії сигнального ліганда протягом тривалого часу, часто втрачають здатність на нього реагувати. Адаптація, або десенсибілізація, зворотна і робить багато клітин особливо чутливими не до абсолютної величини концентрації хімічного сигналу, а до зміни цієї концентрації. Сигнальні ліганди, що приєдналися до поверхневих рецепторів клітин-мішеней, нерідко захоплюються шляхом ендоцитозу, опосередкованого рецепторами. Оскільки ендоцитозні бульбашки зазвичай переносять свій вміст в лізосоми, ліганди, а часто і пов’язаний з ними рецептор розщеплюються гідролітичними ферментами. Цей процес не тільки є головним шляхом розпаду деяких сигнальних лігандів, але і відіграє важливу роль у регулюванні концентрації певних рецепторних білків на поверхні клітин-мішеней. При незвично високих концентраціях сигнальних лігандів, наприклад адреналіну або ацетилхоліну, часто спостерігається інший тип регуляції поверхневих рецепторів. Такі ліганди не викликають ендоцитозу і розщеплювання комплексів ліганд-рецептор, але зворотно інактивують рецептори. Зворотна інактивація поверхневих рецепторів не обов’язково супроводжується втратою здатності зв’язувати ліганд. Довготривале приєднання ацетилхоліну до холінергічних рецепторів м’язової клітини в нервово-м’язовому з’єднанні примушує ці рецептори набувати неактивної конформації, але інактивовані рецептори як і раніше здатні зв’язувати ацетилхолін. Проте інактивовані рецептори не здатні, зв’язавши медіатор, відкривати іонні канали в плазмолемі і викликати потенціал дії. Наприклад, у наркоманів, які вживають морфін, клітини-мішені в мозку десенсибілізовані по відношенню до морфіну, проте мають нормальну кількість опіатних рецепторів. |